前期我们介绍了如何进行chip-qpcr验证,里面提到了一个比较重要的因素——扩增范围的选择及引物的设计。相比双荧光素酶、酵母单杂-点对点验证等允许完整启动子验证的实验,chip-qpcr要求单次验证的范围尽量控制在150-200bp内。但一个基因的启动子一般有2-3k,如果漫无目的地选择区域去验证,不仅花费的成本会很高,检测效果也会大打折扣。在没有进行前期chip-seq的基础上,如何对感兴趣的转录因子(tf)以及靶基因pro进行验证,以及如何筛选靶点,也成为了许多老师困惑的地方。
前期我们介绍了当拥有motif矩阵时,如何利用fimo进行结合位点预测,详见【一文get寻找motif在序列上的定位】。这里,给大家介绍一下如何利用jaspar去寻找感兴趣tf的motif以及利用jaspar-scan进行结合位点预测。
一 「 确定目标转录因子名称与靶基因启动子序列 」
要做验证,当然就要确定好验证对象。当验证对象为转录因子时,大多数老师都会将靶基因的验证区域锁定为启动子序列。那么,我们可以通过ncbi(其他数据库也行,在此以ncbi为例)去找到某个靶基因的启动子序列。我们以stat3(tf)以及cxcl1-pro(靶基因启动子)(物种:human;参考文献:doi:10.1038/s41419-024-06843-y)作为研究对象。核心数据库jaspar core包含的物种类群主要包括vertebrata(脊椎动物),insecta(昆虫),nematoda(线虫),fungi(真菌),plantae(植物)和urochordata(尾索动物)六大类。
(1)cxcl1-pro序列获取
首先在ncbi上找到这个基因的序列,注意看右上角红框部位为这个基因在对应版本基因组染色体上的位置。
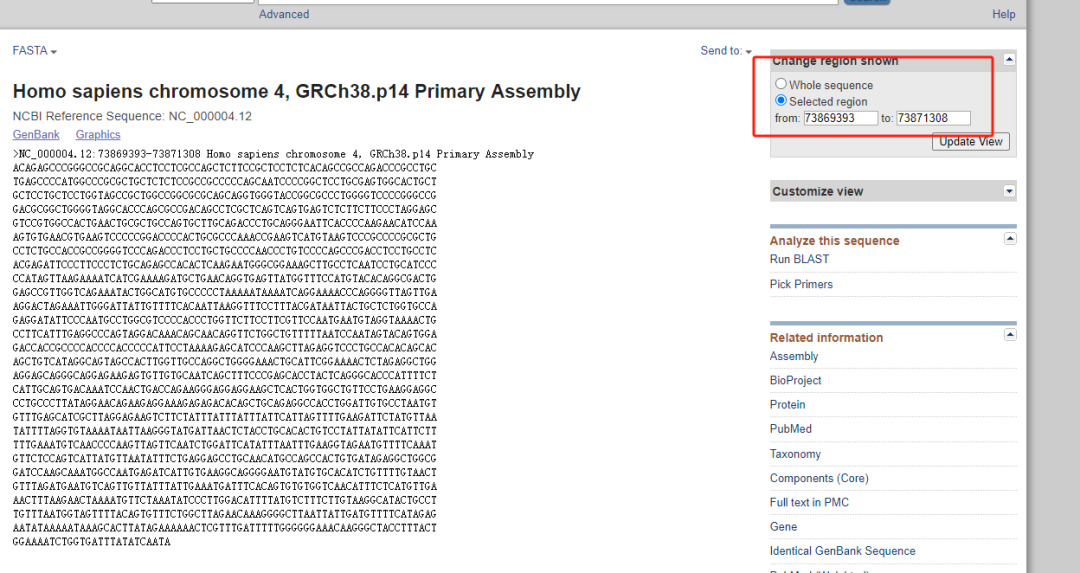
将染色体位置往基因上游调整2kb,即可找到它的启动子序列。这里敲重点:一定要注意这个基因是正链基因还是负链基因!两者启动子序列在染色体上的位置是不同的。例如,cxcl1是正链基因,它在染色质上的位置是nc_000004.12:73869393-73871308。那么上游2kb对应的区域就是nc_000004.12:73867393-73869392。但如果是个负链基因(在这里我们假设cxcl1是个负链基因),其启动子序列则是nc_000004.12:73871309-73873309。

箭头的方向可辅助确认正/负链

cxcl1-pro序列
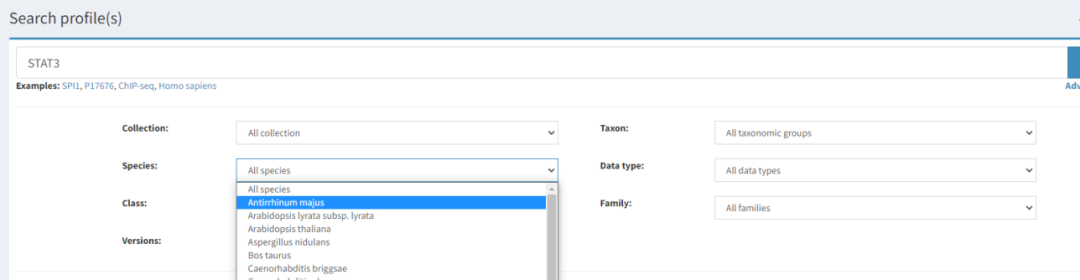
(2)在jaspar找到stat3信息
jaspar网址:https://jaspar.elixir.no/
直接在搜索框中搜索stat3,因为案例物种是人,本文没有额外进行物种的筛选,如果有一些其他的需求,也可以点开advanced options增加筛选条件。
如果出现一个转录因子多个motif,小数点后面的数字越大,表明版本越新(常见的转录因子,不同版本motif展示图可能差异不大,但是实际motif矩阵是不一样的,不同版本motif矩阵预测结果不一定一致)。本文以ma0144.3为例。
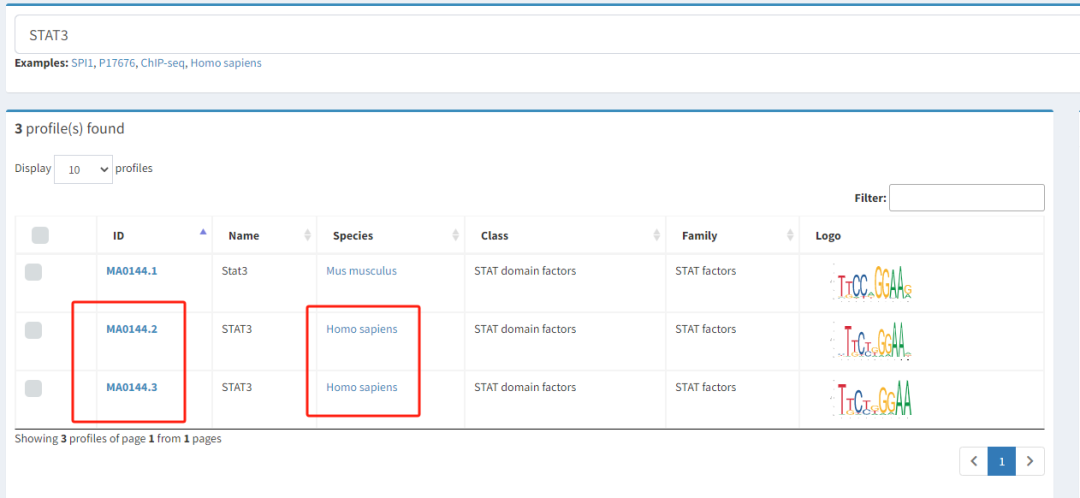
二 「 利用jaspar-scan进行结合位点预测 」
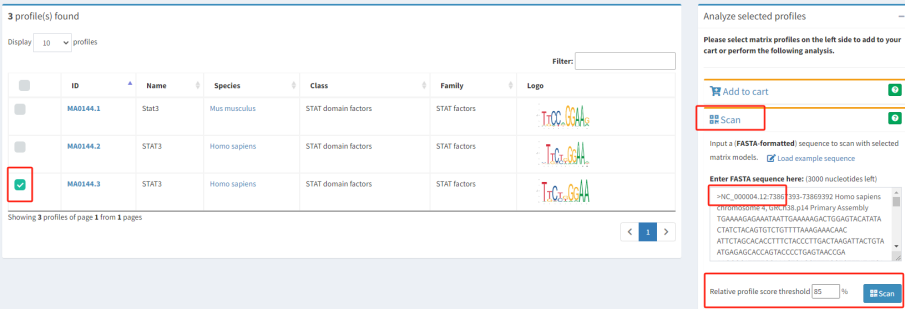
勾选想要分析的版本,点击页面右边scan,将前面找到的cxcl1-pro序列输入进去(如果不是启动子,想分析其他区域序列,请注意输入的序列范围需<3kb)。这里注意不要只输入序列,序列title(>xxx)也需要一起输入才表示这是个fasta格式文件。相对评分阈值(relative profile score threshold)数据库默认为80%,这个值代表数据库认为结合位点的可信度,这里因为结果比较多,我们调整为85%。确认好相关信息后点击scan。
随后的页面就会跳转为预测结果,重点主要关注相对评估值(relative score)以及后面预测的位点。这里以前三为例展示一下在cxcl1-pro中的具体位置。注意,打分排名第二名为负链结合(strand:-),在对应序列时是反向互补配对(dna为双链,motif结合在启动子负链上也是可能的)。
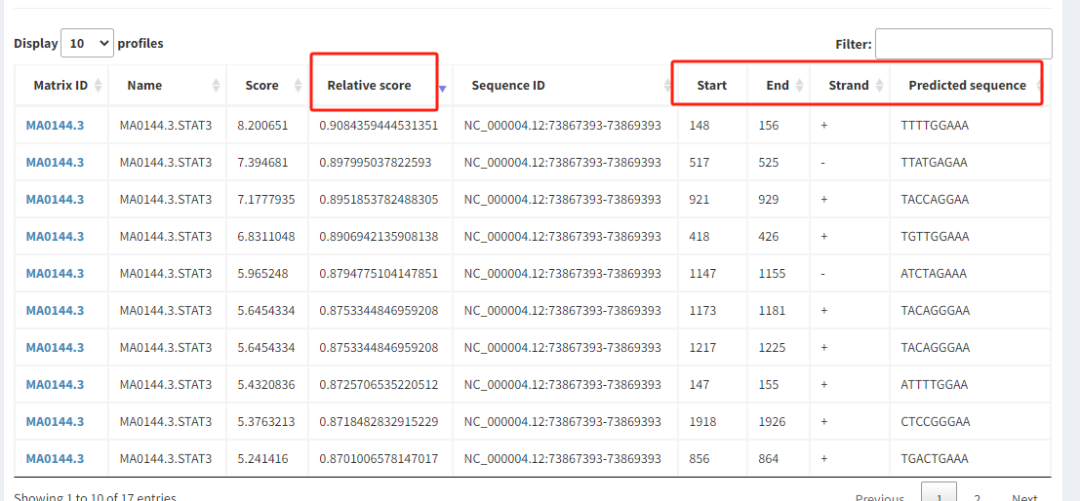

预测打分前三序列定位
这样就做完了结合位点的预测,找到的结合位点就可以用于后期进行chip-qpcr、emsa等需要具体靶点实验的引物/探针设计了。当然,预测结果仅供参考,建议还是同时结合文献、chip测序等结果来挑选位点哦!对chip-qpcr感兴趣的老师也可以参考我们的公众号推文——【如何进行chip-qpcr富集验证】。
发表评论