暑期简单学习了机器学习理论知识,当时跟着b站咕泡老师学的,内容讲得蛮详细,实例代码、资料都比较全面,但是学校python课程开设在这学期,所以用python进行数据分析、建模等不是很熟悉,所以决定用之前学过的r语言来实现机器学习。r语言的相关包也都比较完善,所以想分享一下近期使用r语言实现分类预测建模遇到的问题及解决方法,并且会系统地分享一下几种常见ml二分类方法实现及代码。
数据预处理
我使用的是geo数据库中的乳腺癌转移相关的基因表达谱数据(gse2034、gse1456),前面一个数据集作为训练集,后面一个数据集作为测试集。
我先使用matlab对mat数据文件进行读入,接着进行t检验,筛选出有极显著差异(p<0.01)的基因,对两个数据集的基因作交集(并删除重复基因)得到765个特征,以及一个类标签(1表示预后好,2表示预后差)。
z-score标准化
z-score适用于数据分布过于凌乱、数据的最大值与最小值未知,或者数据中存在过多的离群值的情况。在进行聚类分析、因子分析、主成分分析等多元统计分析时,通常采用这种方法。
在r语言中,可以使用scale函数实现z-score标准化。
gse_train <- read.csv("train_data.csv") #以gse2034为训练集,这里已为预处理后的差异表达数据以及预后label数据
gse_train <- as.data.frame(gse_train)
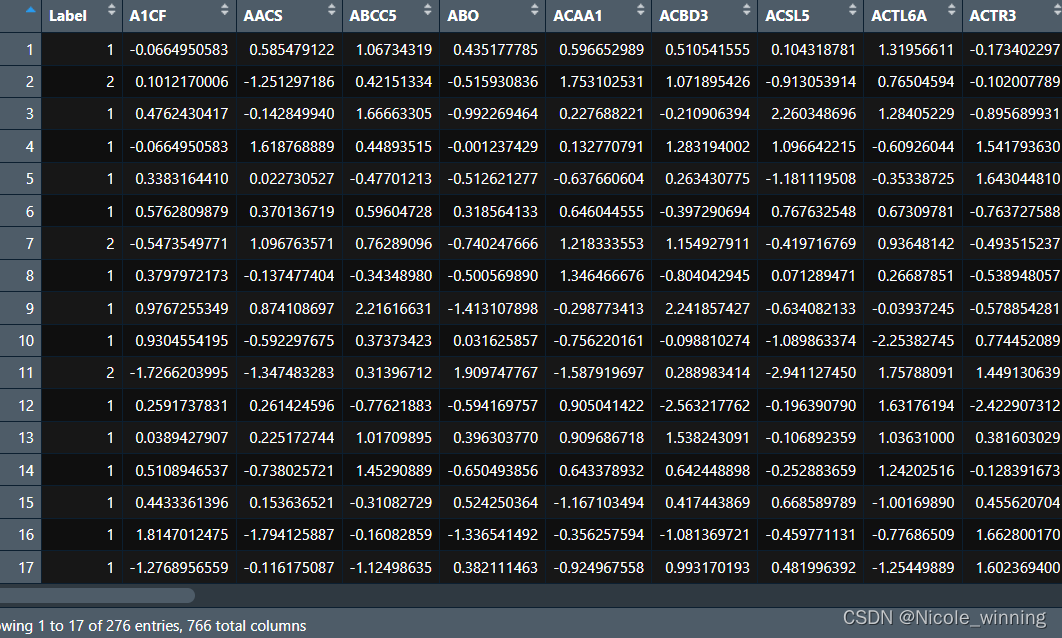
gse_train[,2:766] <- scale(gse_train[,2:766]) #z-score标准化,第一列为类标签不需要标准化
view(gse_train)
gse_test <- read.csv("test_siggene.csv") #以gse1456为测试集,这里已为预处理后的差异表达数据以及预后label数据
gse_test <- as.data.frame(gse_test)
gse_test[,2:766] <- scale(gse_test[,2:766]) #z-score标准化
view(gse_test)下图展示的是gse2034的标准化后的数据
smote处理样本不平衡问题
在这里我注意到,预后标签1和2的数量分布不平衡。在机器学习中,训练数据中不同类别的样本数量分布不均衡可能会导致在训练过程中过多关注数量较多的类别,忽略数量较少的类别,从而使得预测准确率降低。
重采样方法主要有两种,一种是欠采样,即减少数量较多类别的样本数量;另一种是过采样,即增加数量较少类别的样本数量。两种方法各有优劣,这里我使用过采样方法。
| 1(预后好) | 2(预后差) |
| 134 | 59 |
library(dmwr2) #加载相关包
library(smotefamily)
set.seed(123)
sub <- sample(nrow(gse_train),nrow(gse_train)*0.7) #这里不能将所有训练集都进行过采样,取70%或其它
train <- gse_train[sub,]
smote_data <- smote(train[,-1], train[,1]) #使用dmwr2包的smote函数进行过采样
newdata <- smote_data$data #检查经过smote处理后的类别分布
table(newdata$class)进行smote平衡后,样本分布为:
| 1(预后好) | 2(预后差) |
| 134 | 118 |
随机森林(random forest)
模型训练和预测
library(randomforest) #加载相关包
library(proc)
#模型训练
set.seed(123) #设置随机种子数,确保以后再执行代码时可以得到一样的结果
newdata$class <- as.factor(newdata$class) #因变量得为因子型
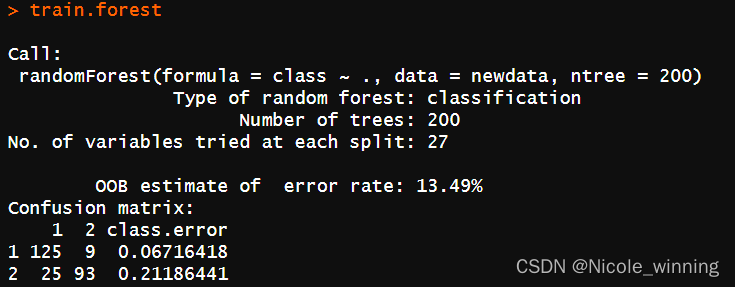
train.forest <- randomforest(class ~ ., data = newdata,ntree = 200) #用平衡后的数据训练
train.forest
查看模型的结果:
分别查看在训练集gse2034和测试集gse1456上的预测结果:
train_probs <- predict(train.forest, gse_train, type = "prob")[, "1"]
test_probs <- predict(train.forest, gse_test, type = "prob")[, "1"]
#计算训练集和测试集的auc并可视化
train_roc <- roc(gse_train$label, train_probs, levels = c("1", "2"))
test_roc <- roc(gse_test$label, test_probs, levels = c("1", "2"))
plot(train_roc, print.auc=true, auc.polygon=true, grid=c(0.1, 0.2),grid.col=c("green", "red"), max.auc.polygon=true,auc.polygon.col="skyblue", print.thres=true,main='rf训练模型roc曲线')
plot(test_roc, print.auc=true, auc.polygon=true, grid=c(0.1, 0.2),grid.col=c("green", "red"), max.auc.polygon=true,auc.polygon.col="skyblue", print.thres=true,main='rf预测模型roc曲线')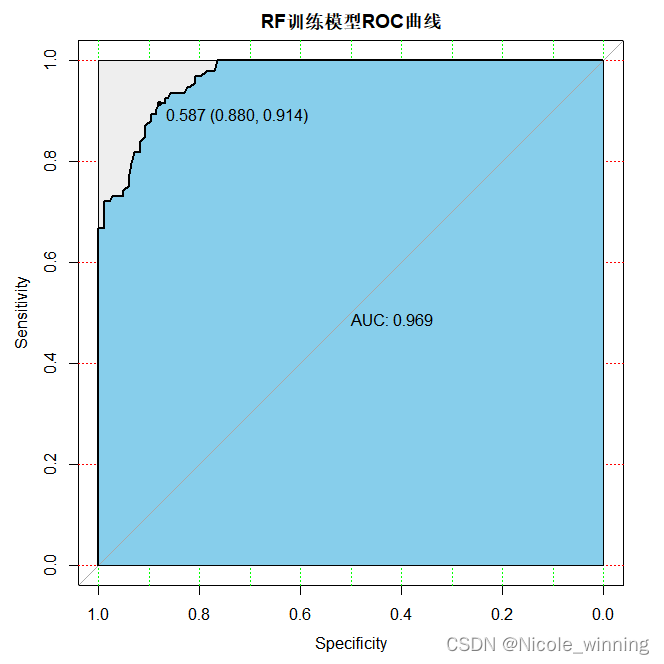
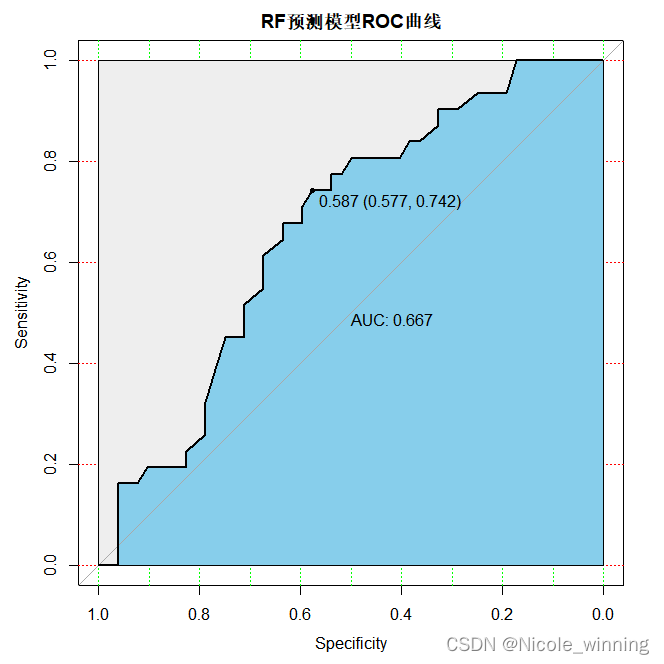
模型评估
常见的评估指标除了auc(roc曲线下面积),还有f1 score值,准确率,召回率等,在这里,我还计算了accuracy = 113/151 = 0.7483
| 预测1 | 预测2 | |
| 实际1 | 112(tn) | 6(fp) |
| 实际2 | 32(fn) | 1(tp) |
变量重要性
importance_gene <- train.forest$importance
head(importance_gene)
varimpplot(train.forest, n.var = min(30, nrow(train.forest$importance)),
main = 'top 30 genes- variable importance') #作图展示 top30 重要的genes
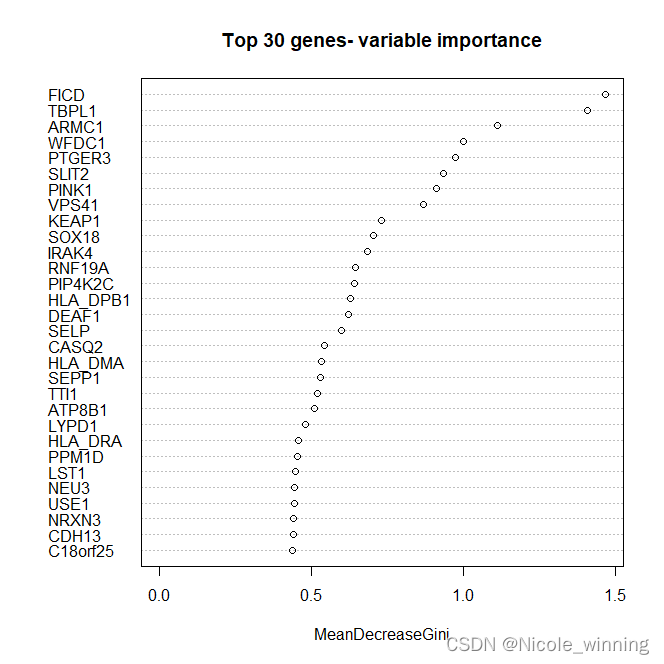
对于上述基因,可以做基因功能注释或基因本体分析(go),以分析这些基因与乳腺癌是否有关系。
支持向量机(support vector machines)
代码编写与上述类似,这里直接展示模型完整代码。
模型训练和预测
#安装并加载相关包
library(e1071)
library(proc)
svm_model <- svm(class~.,data = newdata,kernel = "radial")
summary(svm_model)
##训练集和测试集预测及auc曲线
train_pred <- predict(svm_model,gse_train)
test_pred <- predict(svm_model,gse_test)
train_roc <- roc(gse_train$label, as.numeric(train_pred), levels = c("1", "2"))
test_roc <- roc(gse_test$label, as.numeric(test_pred), levels = c("1", "2"))
plot(train_roc, print.auc=true, auc.polygon=true, grid=c(0.1, 0.2),grid.col=c("green", "red"), max.auc.polygon=true,auc.polygon.col="skyblue", print.thres=true,main='svm训练模型roc曲线')
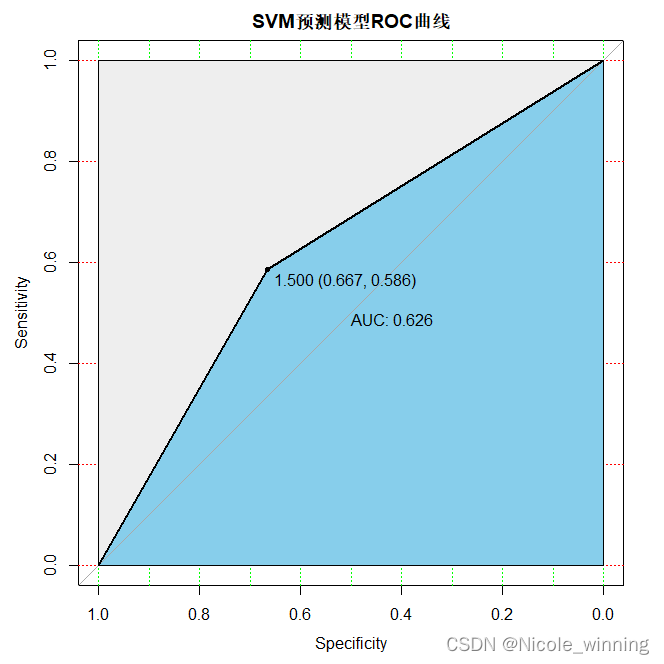
plot(test_roc, print.auc=true, auc.polygon=true, grid=c(0.1, 0.2),grid.col=c("green", "red"), max.auc.polygon=true,auc.polygon.col="skyblue", print.thres=true,main='svm预测模型roc曲线')
svm_model <- svm(class~.,data = newdata,kernel = "radial") #通过改变kernel核函数来建模
summary(svm_model)
svm_pred <- predict(svm_model,gse_train,decision.values = true) #模型预测
tab <- table(predicted = svm_pred,actual = gse_train$label) #展示二联表
tab
1-sum(diag(tab))/sum(tab) #计算错误率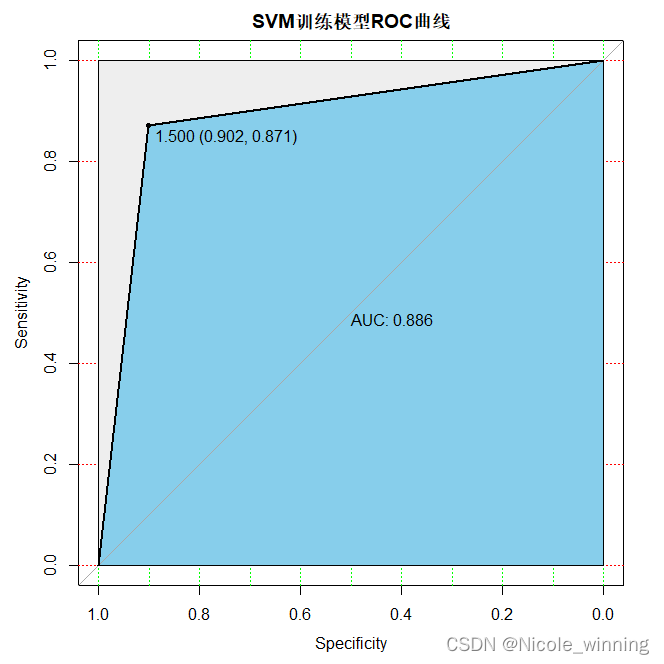
模型评估
svm的auc较随机森林的降低,模型的预测准确性降低,此外,svm的accuracy = 110/151 =0.7285
| 预测1 | 预测2 | |
| 实际1 | 110(tn) | 8(fp) |
| 实际2 | 33(fn) | 0(tp) |
决策树(decision tree)
模型训练和预测
#加载相关包
library(rpart)
library(tibble)
library(bitops)
library(rattle)
library(rpart.plot)
library(rcolorbrewer)
library(rocr)
#构建决策树
set.seed(123)
newdata$class <- as.factor(newdata$class)
dt <- rpart(class ~.,data = newdata)
summary(dt)
#训练集和测试集预测
train.pred <- predict(dt,newdata = gse_train,type = "class")
test.pred <- predict(dt,newdata = gse_test,type = "class")
train_roc <- roc(gse_train$label, as.numeric(train.pred), levels = c("1", "2"))
test_roc <- roc(gse_test$label, as.numeric(test.pred), levels = c("1", "2"))
plot(train_roc, print.auc=true, auc.polygon=true, grid=c(0.1, 0.2),grid.col=c("green", "red"), max.auc.polygon=true,auc.polygon.col="skyblue", print.thres=true,main='dt训练模型roc曲线')
plot(test_roc, print.auc=true, auc.polygon=true, grid=c(0.1, 0.2),grid.col=c("green", "red"), max.auc.polygon=true,auc.polygon.col="skyblue", print.thres=true,main='dt预测模型roc曲线')
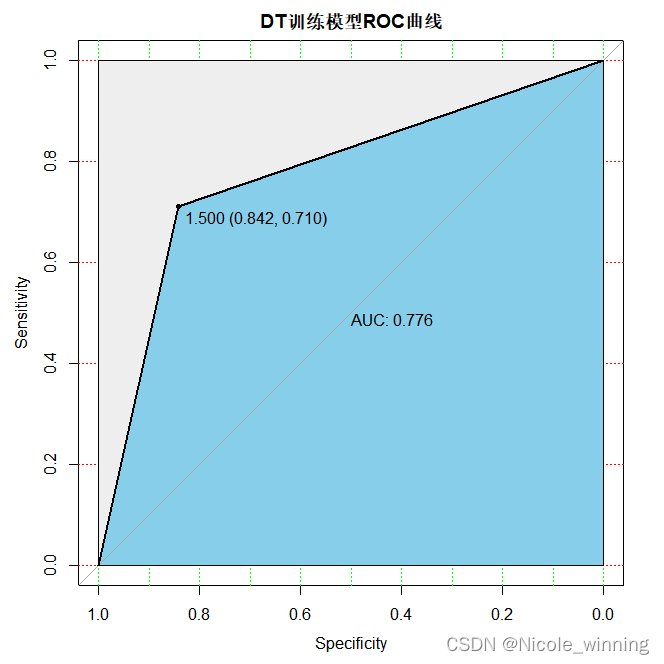
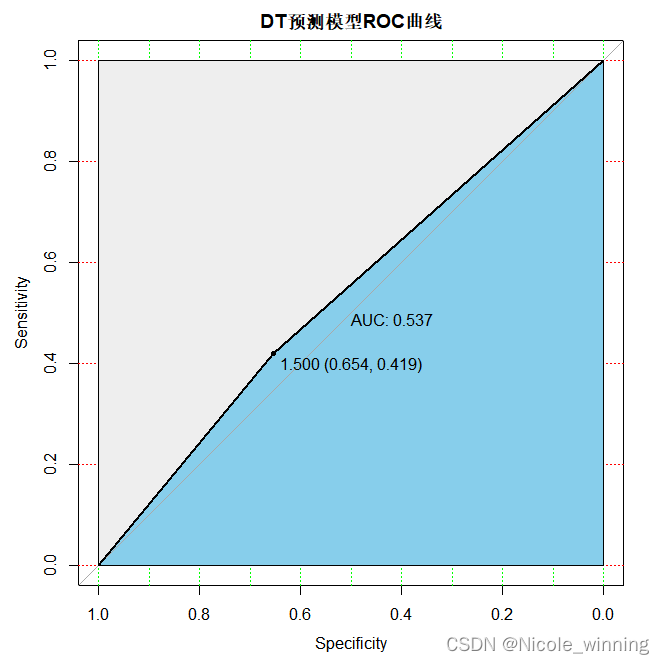
模型评估
从上述roc曲线可以看出,决策树的预测准确性最差。再计算一下准确度,accuracy = 96/151 =0.6358,效果相较前两种比较差。
| 预测1 | 预测2 | |
| 实际1 | 83(tn) | 35(fp) |
| 实际2 | 20(fn) | 13(tp) |
fancyrpartplot(dt) #看一下目前的决策树
plotcp(dt) #画出xerror和cp值与树的节点个数的关系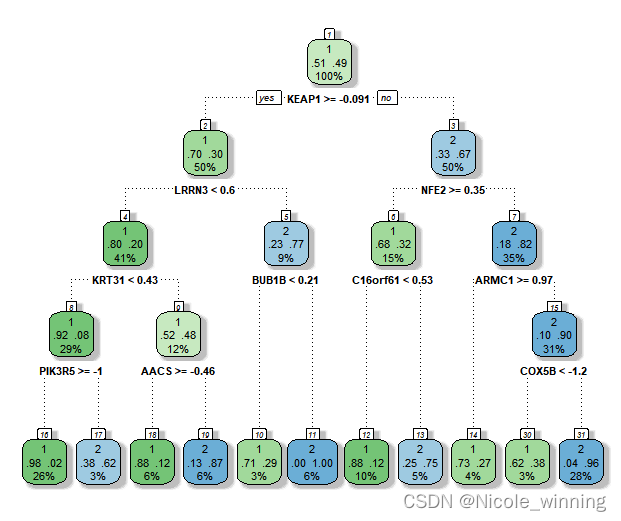
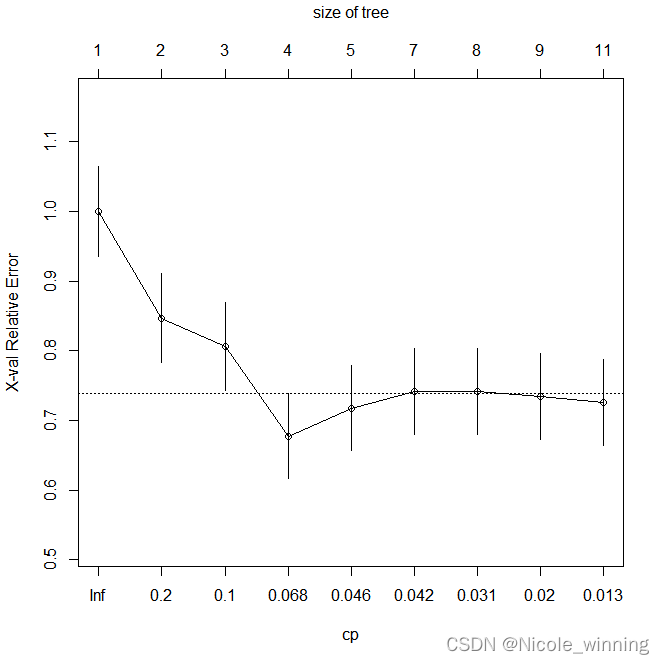
可在此基础上对决策树进行剪枝,选出最优决策树,以期达到更好分类预测的效果。
总结
机器学习建模分析后,还要进行调参或交叉验证以提高模型的预测率,就是所谓的“炼丹”。评估模型也应该用多种指标,包括f1 score,accuracy(准确率),召回率,绘制roc曲线等。此外,机器学习是一个“黑盒子”模型,在得到模型预测结果后,还应该深一步进行生物学解释,由于目前我的生信分析能力还不足,后续学习到了再分享~


发表评论