之前讲过一篇空间转录组的文献,里面首次提出了multimodal intersection analysis(mia)的空间转录组分析思路。

mia分析可以用来评估空间上某个region或者cluster中富集的细胞类型。需要单细胞和空间转录组两种组学数据,数据最好配对。
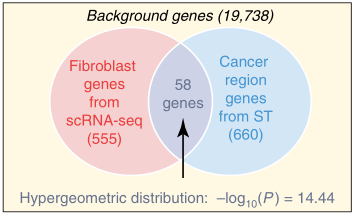
上图是示例图,一个region是否富含某一种细胞类型,看的是一个region高表达的基因和一个celltype高表达的基因是不是有足够多的重叠。
原理不算难,跟常规的「差异基因」做「富集分析」原理是一样的,详细原理之前写过一篇帖子【富集分析的原理与实现】,感兴趣可以看看。
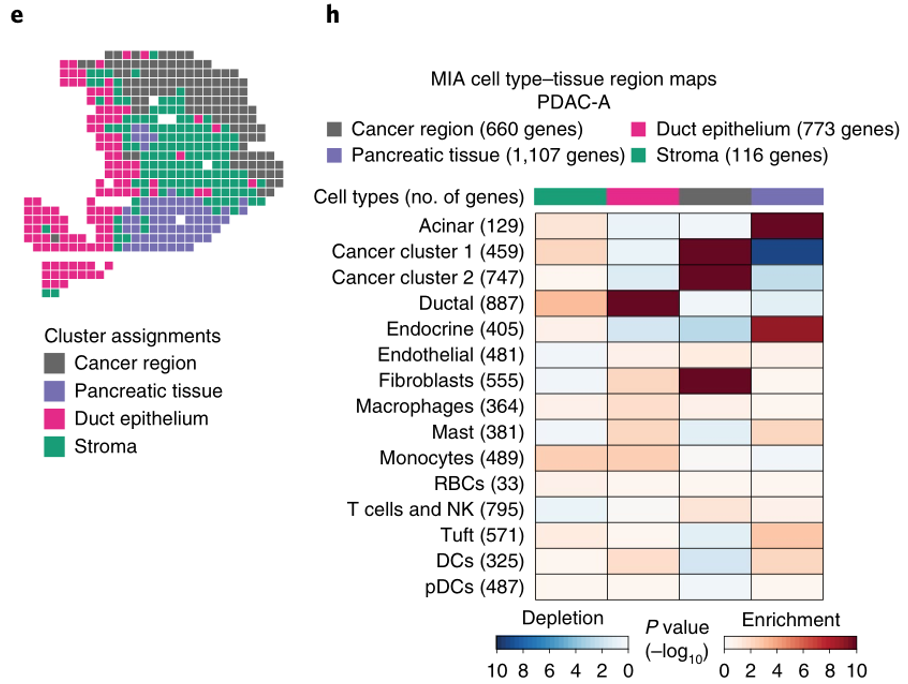
下文的分析会复现这两张小图:原文的fig.2e和fig.2h
1.单细胞数据分析流程
library(seurat)
library(tidyverse)
pdaca.list=readrds("pdac-a.list.rds")
# length(pdaca.list)
# 4
# names(pdaca.list)
# "scrna_count" "scrna_anno" "st_count" "st_coord"
# 该列表包含单细胞count矩阵,单细胞数据集的注释,空间转录组count矩阵,空间转录组spot的坐标
# 由公粽号【top生物信息】整理
### 0.根据原文献fig.2h,将小注释合并 ########################################
pdaca.list$scrna_anno$celltype[str_detect(pdaca.list$scrna_anno$celltype,"^ductal")] = "ductal"
pdaca.list$scrna_anno$celltype[str_detect(pdaca.list$scrna_anno$celltype,"^macrophages")] = "macrophages"
pdaca.list$scrna_anno$celltype[str_detect(pdaca.list$scrna_anno$celltype,"^mdcs")] = "mdcs"
### 1.单细胞转录组流程 #####################################################
pdaca.seu = createseuratobject(counts = pdaca.list$scrna_count)
pdaca.seu <- normalizedata(pdaca.seu, normalization.method = "lognormalize", scale.factor = 10000)
pdaca.seu <- findvariablefeatures(pdaca.seu, selection.method = "vst", nfeatures = 2000)
pdaca.seu <- scaledata(pdaca.seu)
pdaca.seu <- runpca(pdaca.seu, npcs = 50, verbose = false)
### 添加注释信息
pdaca.seu@meta.data$cb=rownames(pdaca.seu@meta.data)
pdaca.seu@meta.data=pdaca.seu@meta.data%>%inner_join(pdaca.list$scrna_anno,by="cb")
rownames(pdaca.seu@meta.data)=pdaca.seu@meta.data$cb
### 降维聚类
pdaca.seu <- findneighbors(pdaca.seu, dims = 1:20)
pdaca.seu <- findclusters(pdaca.seu, resolution = 0.5)
pdaca.seu <- runumap(pdaca.seu, dims = 1:20)
pdaca.seu <- runtsne(pdaca.seu, dims = 1:20)
dimplot(pdaca.seu,group.by = "celltype",reduction = "umap",pt.size = 1,label = t,repel = t,label.size = 4)
### 找差异基因
# 控制三个阈值:logfc.threshold p_val_adj d
idents(pdaca.seu)="celltype"
maintype_marker=findallmarkers(pdaca.seu,logfc.threshold = 0.5,only.pos = t)
maintype_marker=maintype_marker%>%filter(p_val_adj < 1e-05)
maintype_marker$d=maintype_marker$pct.1 - maintype_marker$pct.2
maintype_marker=maintype_marker%>%filter(d > 0.2)
maintype_marker=maintype_marker%>%arrange(cluster,desc(avg_log2fc))
maintype_marker=as.data.frame(maintype_marker)
2.空间转录组流程
注意:
-
这个文献只能下载到空转矩阵和spot坐标,没有图像。因此下面的流程和单细胞转录组一模一样; -
若是完整的空转数据集,则有一些步骤是不一样的,具体流程可以参考seurat官网,https://satijalab.org/seurat/articles/spatial_vignette.html
pdaca.v.seu = createseuratobject(counts = pdaca.list$st_count)
pdaca.v.seu <- sctransform(pdaca.v.seu, verbose = false)
pdaca.v.seu <- runpca(pdaca.v.seu, assay = "sct", verbose = false)
pdaca.v.seu <- findneighbors(pdaca.v.seu, reduction = "pca", dims = 1:30)
pdaca.v.seu <- findclusters(pdaca.v.seu, verbose = false)
pdaca.v.seu <- runumap(pdaca.v.seu, reduction = "pca", dims = 1:30)
pdaca.v.seu <- runtsne(pdaca.v.seu, reduction = "pca", dims = 1:30)
### 整合坐标、region
#(此处把坐标类比单细胞转录组分析中的注释信息)
# region信息由公粽号【top生物信息】整理
region.info=read.table("pdac_a.region.txt",header = t,sep = "\t",stringsasfactors = f)
pdaca.v.seu@meta.data$sb=rownames(pdaca.v.seu@meta.data)
pdaca.v.seu@meta.data=pdaca.v.seu@meta.data%>%inner_join(region.info,by = "sb")
rownames(pdaca.v.seu@meta.data)=pdaca.v.seu@meta.data$sb
### 画图看看
library(rcolorbrewer)
library(scales)
color_region=c("#666666","#766fb1","#e42a88","#189d77")
names(color_region)=c("cancer region","pancreatic tissue","duct epithelium","stroma")
pdaca.v.seu@meta.data%>%ggplot(aes(x=x_coord,y=y_coord,fill=region))+
geom_tile(color="white")+
scale_fill_manual("cluster assignments",values = color_region)+
theme_void()+
theme(
legend.title = element_text(size = 16),
legend.text = element_text(size = 14),
legend.position = "bottom",
legend.direction = "vertical"
)+
guides(fill = guide_legend(override.aes = list(size=10)))
ggsave("fig.2e.pdf",width = 10,height = 14,units = "cm")
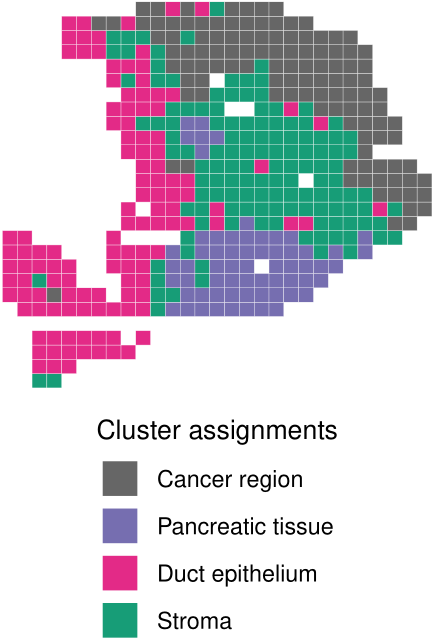
### 找region特异基因
idents(pdaca.v.seu)="region"
region_marker=findallmarkers(pdaca.v.seu,logfc.threshold = 0,only.pos = t)
region_marker=region_marker%>%filter(p_val_adj < 0.1)
region_marker$d=region_marker$pct.1 - region_marker$pct.2
region_marker=region_marker%>%filter(d > 0.05)
region_marker=region_marker%>%arrange(cluster,desc(avg_log2fc))
region_marker=as.data.frame(region_marker)
说明:
-
1.上述找两个deg数据框的方法不唯一,阈值也不唯一 -
2.第二个deg数据框也可以是空间cluster的marker -
3.mia分析模式在单细胞和空间转录组场景都可以应用,空转场景是看细胞亚群的富集程度,单细胞场景是做细胞亚群注释
3.mia分析
region_specific=region_marker[,c("cluster","gene")]
colnames(region_specific)[1]="region"
celltype_specific=maintype_marker[,c("cluster","gene")]
colnames(celltype_specific)[1]="celltype"
n=length(union(rownames(pdaca.seu),rownames(pdaca.v.seu)))
library(rcolorbrewer)
library(scales)
color_region=c("#666666","#766fb1","#e42a88","#189d77")
names(color_region)=c("cancer region","pancreatic tissue","duct epithelium","stroma")
source("symia.r")
miares=symia(region_specific,celltype_specific,n,color_region)
之后会返回一个数据框,以及自动生成一张图
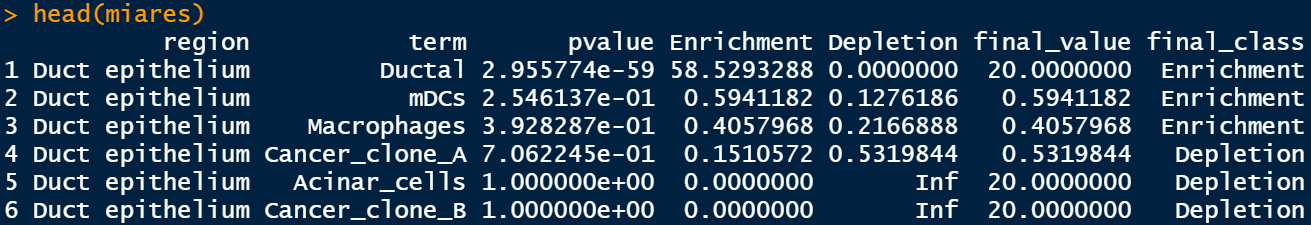
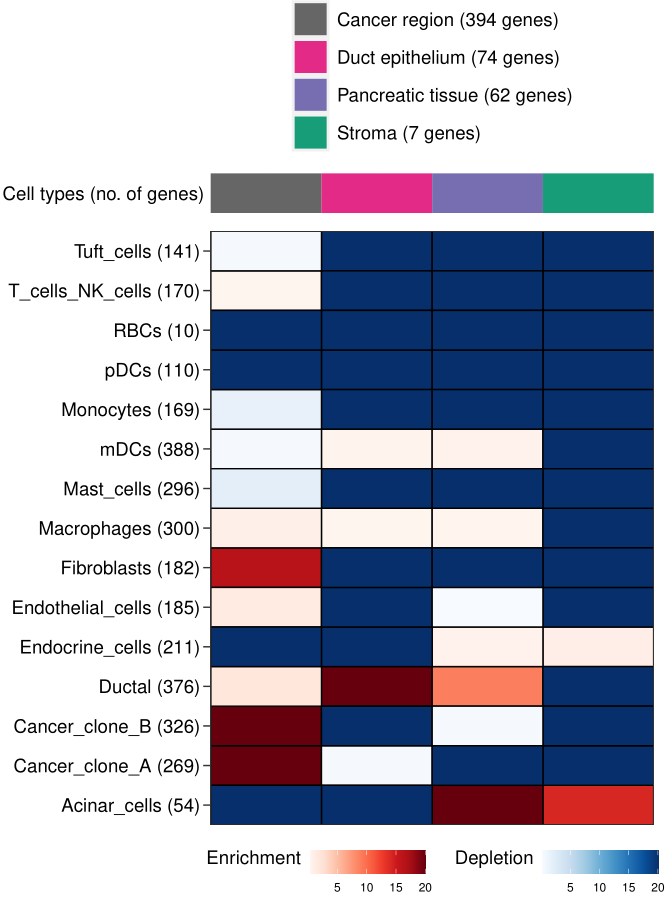
可以看到cancer region同时富集了cancer cell和fibroblast,这和文章结果是「一致」的。
当然上图和原文有些差别,原因是我没有采用原文找差异基因的方法,而是选用了seurat中的常规方法,这个灵活选择。
本文数据整理和代码编写花费大量时间,故不无偿提供,有需要的朋友可以关注公粽号,然后回复2022b



发表评论