metaphlan 4是一种基于dna序列的微生物组分析工具,它能够从宏基因组测序数据中识别和分离微生物的组成。以下是安装和使用metaphlan 4的步骤:
安装metaphlan 4:
先看原站点和仓库,暂时访问不了的可以隔一段时间再试:
metaphlan4 – the huttenhower lab
裸机环境,手动安装
1. 安装依赖项:
metaphlan 4需要python 3.7以上的版本(建议使用anaconda环境),同时还需要安装biopython、pandas和numpy等包。可以使用pip命令进行安装,例如:
pip install biopython pandas numpy
2. 下载metaphlan 4程序:
从metaphlan 4的官方网站(https://github.com/biobakery/metaphlan)下载最新的metaphlan 4程序,并解压缩到指定目录中。
数据库地址(建议手动下载,地址及文件见下方)
要配置metaphlan4最新数据库,您可以按照以下步骤进行操作:
1. 下载最新的metaphlan4数据库文件,可以在metaphlan4官方网站上下载。
2. 解压缩下载的metaphlan4数据库文件,得到一个包含多个文件的目录。
3. 打开metaphlan4配置文件,通常为"metaphlan_database.cfg",可以在metaphlan4的安装目录中找到。
4. 在配置文件中,找到"bowtie2db"和"mpa_pkl"这两个参数,并将它们的值分别改为metaphlan4数据库目录下的"bowtie2"和"mpa"文件的路径。
5. 保存配置文件。
使用conda环境安装(推荐)
本示例使用anaconda3,
以下是在anaconda3中安装metaphlan4的步骤:
步骤1:安装conda
如果您尚未安装conda,请使用以下命令在终端中安装:
到这里去找安装包吧,什么版本都有,这里下载linux64最新版
https://repo.anaconda.com/archive
###下载安装包
wget https://repo.anaconda.com/archive/anaconda3-2023.09-0-linux-x86_64.sh
###安装
sh anaconda3-2023.09-0-linux-x86_64.sh
###先进去后会要让同意许可协议,注意太快可能错过跳出,到后面慢一点向下翻
#到最后询问是否同意时输入yes
##遇到按键太快跳过的话重新执行安装命令再次进入
#在配置安装目录时要注意输入自己想要安装的目录
步骤2:创建conda环境
打开终端并输入以下命令,创建一个名为“metaphlan4”的conda环境:
#创建metaphlan4的conda环境
conda create -n metaphlan4 python=3.7
mamba create -n metaphlan4 python=3.7步骤3:激活conda环境
输入以下命令,激活“metaphlan4”环境:
#激活指定环境,任选一种,建议mamba
conda activate metaphlan4
mamba activate metaphlan4
source activate metaphlan4步骤4:安装metaphlan4
使用以下命令安装metaphlan4:
#在激活环境中安装metaphlan
conda install -c bioconda -c conda-forge metaphlan
#mamba也挺不错的,速度还快,自己配置即可
mamba install -c bioconda -c conda-forge metaphlan
#需要一点时间,等待完成步骤5:测试metaphlan4
输入以下命令测试安装是否成功:
metaphlan --version
如果可以正确显示版本信息,则安装成功。
步骤6:配置metaphlan数据库
默认情况下可以使用metaphlan --install命令可以安装数据库,按照官方建议添加--bowtie2db参数,并指定conda目录外的专用数据库目录/mnt/metaphlan4_db,大家按需求修改。指定conda外目录的好处就是conda环境目录容量小一些,重新安装时还可以设置此目录作为数据库目录就行,不用重新下载和配置了。
metaphlan --install --bowtie2db /mnt/metaphlan4_db
# metaphlan自动下载的数据库有可能不是最新的,大家可以清空指定文件夹,
# 或将已有数据文件全部移到其他备份目录
# 然后编辑一个mpa_latest
cd /mnt/metaphlan4_db
vim mpa_latest
mpa_vjun23_chocophlansgb_202307
## 这个文件不要有空格不要有空行,就单独这个版本的名称独占一行即可。
可下载版本见ftp: index of /biobakery4/metaphlan_databases
写入mpa_latest文件中的仅如下面红线标注的版本名称,都是日期结尾
mpa_vjun23_chocophlansgb_202307
mpa_voct22_chocophlansgb_202212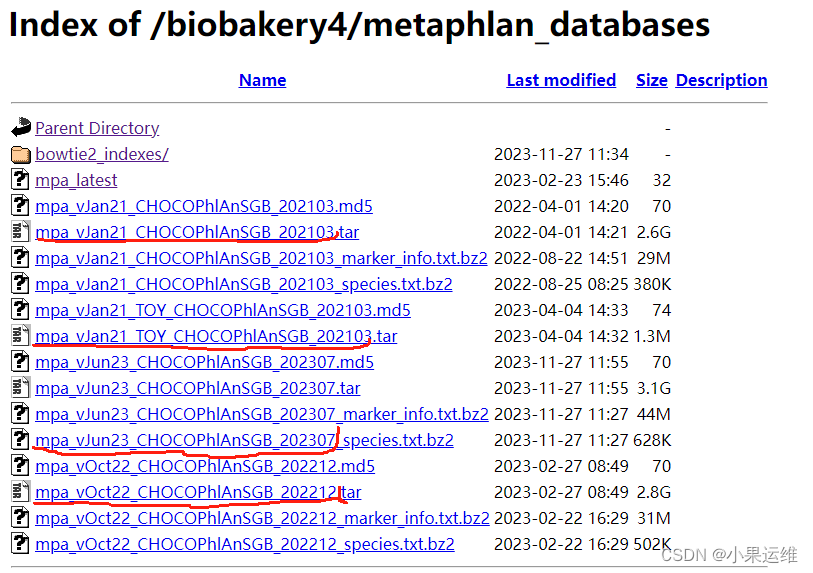
开始后会自动下载文件和运行bowtie索引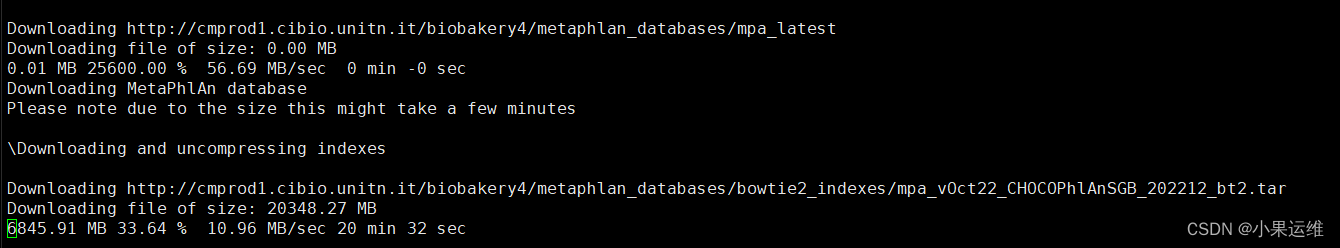
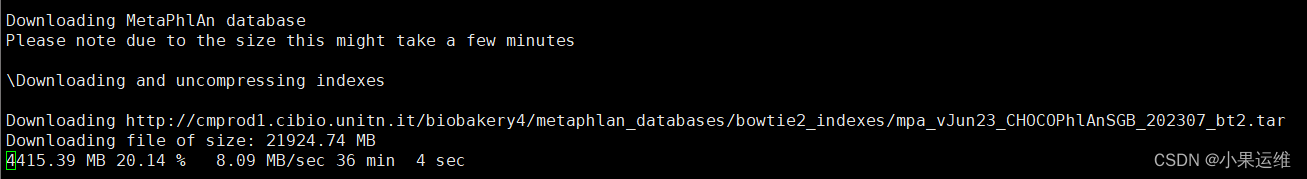
有时候下载速度慢,可能引起失败,建议手动下载最新数据库:
地址在这里:http://cmprod1.cibio.unitn.it/biobakery4/metaphlan_databases/
下载这几个文件:
http://cmprod1.cibio.unitn.it/biobakery4/metaphlan_databases/bowtie2_indexes/mpa_voct22_chocophlansgb_202212_bt2.tar http://cmprod1.cibio.unitn.it/biobakery4/metaphlan_databases/mpa_voct22_chocophlansgb_202212.md5
http://cmprod1.cibio.unitn.it/biobakery4/metaphlan_databases/mpa_voct22_chocophlansgb_202212.tar
http://cmprod1.cibio.unitn.it/biobakery4/metaphlan_databases/mpa_latest
下载完成后将所有下载文件放入下面目录(根据自己安装目录找ananconda3的位置,后面路径都一样):
anaconda3/envs/mpa/lib/python3.7/site-packages/metaphlan/metaphlan_databases
后面第一次去运行metaphlan去注释你的序列的时候会自动建库
使用metaphlan 4:
1. 准备输入文件:metaphlan 4需要输入fastq或fasta格式的文件。如果是pe读取,需要将两个序列文件合并为一个文件。
2. 运行metaphlan 4:在终端中进入metaphlan 4的安装目录,并运行以下命令:
python3 metaphlan --input_file example.fastq --output_file output.txt其中,--input_file参数指定输入文件的路径和名称,--output_file参数指定输出文件的路径和名称。
3. 查看结果:metaphlan 4的输出文件包含了每个微生物的相对丰度和物种注释信息。可以使用文本编辑器或者excel等软件打开输出文件进行查看和分析。
conda环境下使用metaphlan4
##激活conda环境
source activate metaphlan4
#直接使用压缩包文件运行,建议使用nohup运行,因为运行时间比较长
#nohup
nohup metaphlan f1.fastq.gz,r2.fastq.gz --bowtie2out f1r2.bowtie2.bz2 --nproc 60 --input_type fastq >f1r2_mtphlan.txt 2>&1 &
#直接运行
metaphlan f1.fastq.gz,r2.fastq.gz --bowtie2out f1r2.bowtie2.bz2 --nproc 60 --input_type fastq -o f1r2_mtphlan.txt
####其中 f1.fastq.gz和r2.fastq.gz分别为样品的双端序列的两个压缩文件,最终我们想要的是f1r2_mtphlan.txt结果解释:
###前面几行已被注释,使用metaphlan工具合并时会自动过滤掉
#anaconda3/envs/metaphlan4/bin/metaphlan 1.fastq,2.fastq --bowtie2out 1.bt2.bz2 --nproc 30 --input_type fastq -o 1.profiled.txt
#76553269 reads processed
#sampleid metaphlan_analysis
#clade_name ncbi_tax_id relative_abundance additional_species
k__bacteria 2 99.92666
k__archaea 2157 0.07334
k__bacteria|p__proteobacteria 2|1224 89.13284
k__bacteria|p__actinobacteria 2|201174 8.93442
k__bacteria|p__bacteroidetes 2|976 1.83546
私房菜:
###使用merge_metaphlan_tables.py将所有样品的注释结果合并(需要激活metaphlan4的conda环境)
merge_metaphlan_tables.py *.txt > merged_abundance_table.txt
###使用下面语句从合并表中提取物种种水平下的物种注释信息
grep -e '(s__)|(clade_name)' merged_abundance_table.txt |grep -v 't__'|sed 's/^.*s__//g'|awk '{$2=null;print}'|sed 's/\ \ /\ /g'|sed 's/\ /\t/g' > merged_abundance_species.txt
###使用下面语句从合并表中提取物种属水平下的物种注释信息
grep -e '(g__)|(clade_name)' merged_abundance_table.txt |grep -v 's__'|sed 's/^.*g__//g'|awk '{$2=null;print}'|sed 's/\ \ /\ /g'|sed 's/\ /\t/g' > merged_abundance_genus.txt
###使用下面语句从合并表中提取物种科水平下的物种注释信息
grep -e '(f__)|(clade_name)' merged_abundance_table.txt |grep -v 'g__'|sed 's/^.*f__//g'|awk '{$2=null;print}'|sed 's/\ \ /\ /g'|sed 's/\ /\t/g' > merged_abundance_family.txt
###使用下面语句从合并表中提取物种目水平下的物种注释信息
grep -e '(o__)|(clade_name)' merged_abundance_table.txt |grep -v 'f__'|sed 's/^.*o__//g'|awk '{$2=null;print}'|sed 's/\ \ /\ /g'|sed 's/\ /\t/g' > merged_abundance_order.txt
###使用下面语句从合并表中提取物种纲水平下的物种注释信息
grep -e '(c__)|(clade_name)' merged_abundance_table.txt |grep -v 'o__'|sed 's/^.*c__//g'|awk '{$2=null;print}'|sed 's/\ \ /\ /g'|sed 's/\ /\t/g' > merged_abundance_class.txt
###使用下面语句从合并表中提取物种门水平下的物种注释信息
grep -e '(p__)|(clade_name)' merged_abundance_table.txt |grep -v 'c__'|sed 's/^.*p__//g'|awk '{$2=null;print}'|sed 's/\ \ /\ /g'|sed 's/\ /\t/g' > merged_abundance_phylum.txt这里有最新2023关于metaphlan分析的workshop内容,大家可以先看一下,基本与上面内容一致。
metaphlan workshop on genomics 2023 · biobakery/metaphlan wiki · github
注意事项:
1. metaphlan 4需要消耗大量的计算资源,建议在具有足够内存和计算能力的计算机上运行。
2. metaphlan 4的结果可能会受到样本质量、参考基因组库的完整性和准确性等多种因素的影响。可以根据需要选择合适的参考基因组库进行分析,以获得更准确的结果。
metaphlan分析结果的使用建议:
1. 了解metaphlan输出格式:metaphlan输出文件包括两个文件,即<filename>.txt和<filename>.txt.bak。其中,<filename>.txt文件是包含分类结果和相对丰度值的文本文件,<filename>.txt.bak文件是二进制文件存储的序列信息。
2. 解释metaphlan输出结果:metaphlan输出文件提供了各个分类单元的相对丰度值,这些单元包括细菌、古菌、真菌和叶绿体等。通过观察这些相对丰度值,可以了解样本中各个分类单元的富集度和活跃度。
3. 与其他工具结合使用:metaphlan结果可以与其他宏基因组分析工具结合使用,如picrust、stamp和lefse等,这些工具可以进一步分析样本中的生物学差异。
4. 结果可视化:metaphlan的结果可以用不同的可视化工具展示,如circos、r、stamp和phinch等。这些工具可以帮助用户更直观地理解样品之间的差异以及分类单元的富集度。
总之,metaphlan是一个强大的宏基因组分析工具,可以帮助用户快速了解样品中的微生物组成。将其结果与其他工具结合使用,可以更深入地挖掘样品差异,为后续的实验设计提供重要参考。
关于宏基因组学相关分析大家还可以参考下面这些脚本和分析流程:
 https://blog.csdn.net/zrc_xiaoguo/article/details/134871717?ops_request_misc=%257b%2522request%255fid%2522%253a%2522170200580516800185874313%2522%252c%2522scm%2522%253a%252220140713.130102334.pc%255fblog.%2522%257d&request_id=170200580516800185874313&biz_id=0&utm_medium=distribute.pc_search_result.none-task-blog-2~blog~first_rank_ecpm_v1~rank_v31_ecpm-3-134871717-null-null.nonecase&utm_term=metaphlan&spm=1018.2226.3001.4450
https://blog.csdn.net/zrc_xiaoguo/article/details/134871717?ops_request_misc=%257b%2522request%255fid%2522%253a%2522170200580516800185874313%2522%252c%2522scm%2522%253a%252220140713.130102334.pc%255fblog.%2522%257d&request_id=170200580516800185874313&biz_id=0&utm_medium=distribute.pc_search_result.none-task-blog-2~blog~first_rank_ecpm_v1~rank_v31_ecpm-3-134871717-null-null.nonecase&utm_term=metaphlan&spm=1018.2226.3001.4450



发表评论