tcga 数据分析实战 —— gsva、ssgsea 和单基因富集分析
前言
前面,我们介绍过了差异基因的功能富集分析,今天,我们对这部分的内容作一些补充
主要介绍一下 geva、ssgsea 和单基因的富集分析
gsva
我们知道,gsea 富集分析方法是针对两组样本来进行评估的,也就是说对基因列表的排列方式是根据基因与表型的相关度(例如,fc 值)来计算的,无法对单个样本使用
其富集分数(enrichment score,es)的计算方式为

依次判断基因列表中的基因是否在基因集合中,如果在基因集合中,则 es 加上该基因与表型的相关度,如果不是集合中的基因,则减去对应的值,最后可以计算出一个最大 es。
gene set variation analysis(gsva)与 gsea 的原理类似,只是计算每个基因集合在每个样本中的 enrichment statistic(es,或 gsva score),其算法流程如下

不同于 gsea 之处在于,对于不同的数据类型(只支持 log 表达值或原始的 read counts 值),假设了不同的累积密度函数(cumulative density function,cdf)
- 芯片数据:正态分布密度函数
rna-seq数据:泊松分布密度函数
而且,gsva 是为每个样本的每个基因计算对应的 cdf 值,然后根据该值对基因进行排序,这样,每个样本都有一个从大到小排序的基因列表
对于某一基因集合,计算其在每个样本中的 es 值,也就是评估基因集合在基因列表中的富集情况。
例如,我们有一个排序后的样本

基因集合包含:b、e、h,我们可以绘制这样一张 k-s 分布图

x 轴为排序后的基因顺序,依照这一顺序,如果基因在集合内,则累积和会加上该基因的值(与基因的顺序有关,排名越靠前值越大),否则累积和不变。
将基因列表分为基因集内核基因集外两个集合,就可以绘制两个分布(红色和绿色曲线),分别计算两个分布之间的最大间距,以基因集内的分布值更大视为正间距(即红色曲线更高),两个最大间距之和即为该基因集的 es 值
这样,就把基因水平的表达矩阵转换成了基因集水平的评分矩阵,可以使用差异表达基因识别算法,寻找显著差异的基因集,从而达到类似功能富集的作用
gsva 分析
先获取基因表达矩阵,我们使用 tcga 肺腺癌和肺鳞癌各 10 个样本的 read counts 数据
library(tcgabiolinks)
get_prep <- function(cancer) {
query <- gdcquery(
project = cancer,
data.category = "transcriptome profiling",
data.type = "gene expression quantification",
workflow.type = "star - counts",
sample.type = c("primary tumor"),
)
# 选择 20 个样本
query$results[[1]] <- query$results[[1]][1:20,]
gdcdownload(query)
# 获取 read count
exp.count <- gdcprepare(
query,
summarizedexperiment = true,
)
return(exp.count)
}
luad.count <- get_prep("tcga-luad")
lusc.count <- get_prep("tcga-lusc")
dataprep_luad <- tcgaanalyze_preprocessing(
object = luad.count,
cor.cut = 0.6,
datatype = "unstranded"
)
dataprep_lusc <- tcgaanalyze_preprocessing(
object = lusc.count,
cor.cut = 0.6,
datatype = "unstranded"
)
# 合并数据并使用 gccontent 方法进行标准化
datanorm <- tcgaanalyze_normalization(
tabdf = cbind(dataprep_luad, dataprep_lusc),
geneinfo = tcgabiolinks::geneinfoht,
method = "gccontent"
)
# 分位数过滤
datafilt <- tcgaanalyze_filtering(
tabdf = datanorm,
method = "quantile",
qnt.cut = 0.25
)
gene.counts <- as.data.frame(datafilt) %>%
tibble::rownames_to_column("gene_id") %>%
inner_join(dplyr::select(tcgabiolinks::get.grch.biomart("hg38"), gene_id, gene_name)
%>% mutate(gene_id = str_remove(gene_id, "\\.\\d+"))) %>%
dplyr::select(-gene_id) %>%
group_by(gene_name) %>%
summarise_all(mean) %>%
tibble::column_to_rownames(var = "gene_name") %>%
rename_with(function(x) substr(x, 1, 12))
# 将数据拆分
luad.exp <- dplyr::select(gene.counts, substr(luad.count$barcode, 1, 12))
lusc.exp <- dplyr::select(gene.counts, substr(lusc.count$barcode, 1, 12))
我们使用 gsva 包提供的 gsva 函数来将基因表达矩阵转换为基因集分数矩阵
library(gsva)
library(gseabase)
# 读取从 gsea 官网下载的通路数据
c2gmt <- getgmt("~/downloads/data/pathway/c2.cp.v7.2.symbols.gmt")
# 删选出常用的这三个数据库中的通路
gene.set <- c2gmt[grep("^kegg|reactome|biocarta", names(c2gmt)),]
# gsva 分析,read counts 使用泊松分布,通路至少包含 10 个基因
gs.exp <- gsva(as.matrix(gene.counts), gene.set, kcdf = "poisson", min.sz = 10)
虽然 gsvadata 包提供了通路数据 c2broadsets 是基因 id,但我们的基因表达数据的行是基因 symbol,所以通路信息也必须是 symbol 格式,要进行格式转换,比较麻烦
所以我们使用 gseabase 包提供的 getgmt 函数来读取从 gsea 官网下载的 c2 通路信息
得到结果如下,共包含 1642 条通路
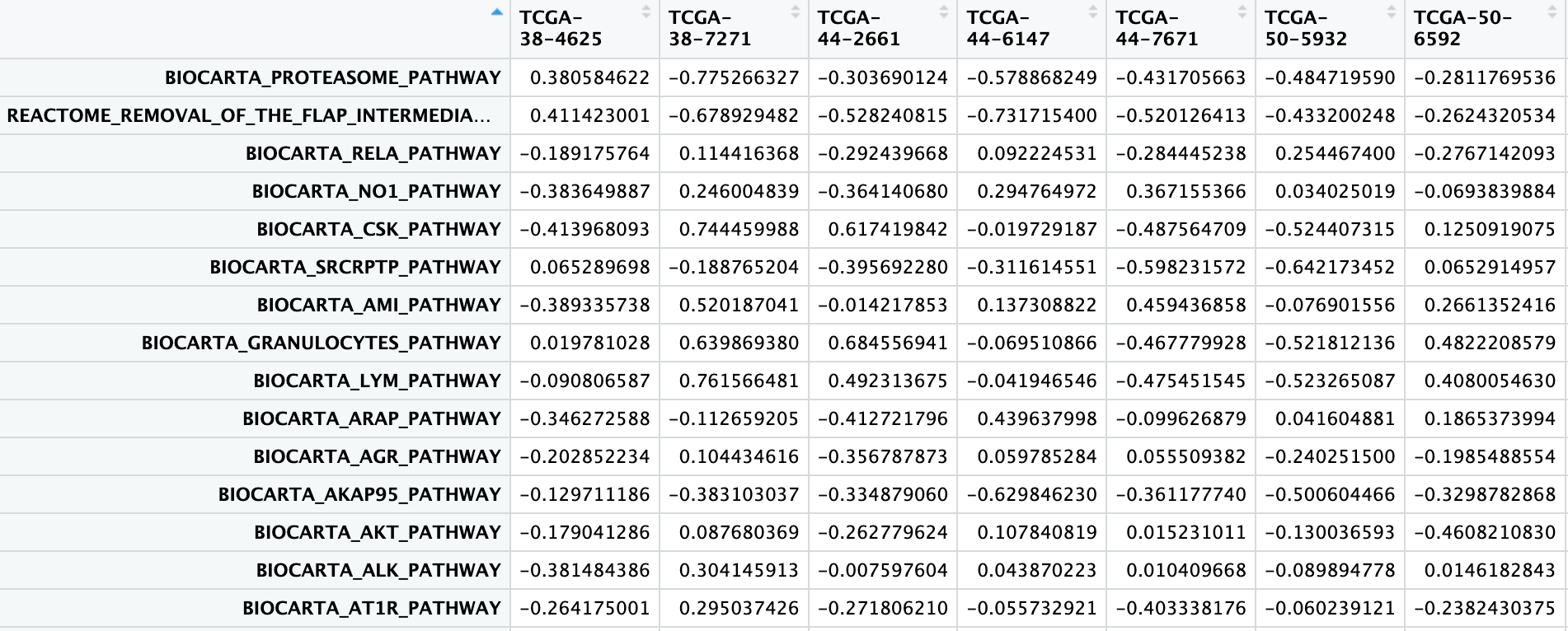
然后,使用差异基因识别方法
差异分析
我们使用 limma 分析差异通路
dea.gs <- tcgaanalyze_dea(
mat1 = gs.exp[, colnames(luad.exp)],
mat2 = gs.exp[, colnames(lusc.exp)],
metadata = false,
pipeline = "limma",
cond1type = "luad",
cond2type = "lusc",
fdr.cut = 0.05,
logfc.cut = 0.5,
)
通过设置 fdr = 0.05,logfc = 0.5 共筛选出 13 条差异通路

查看通路的火山图
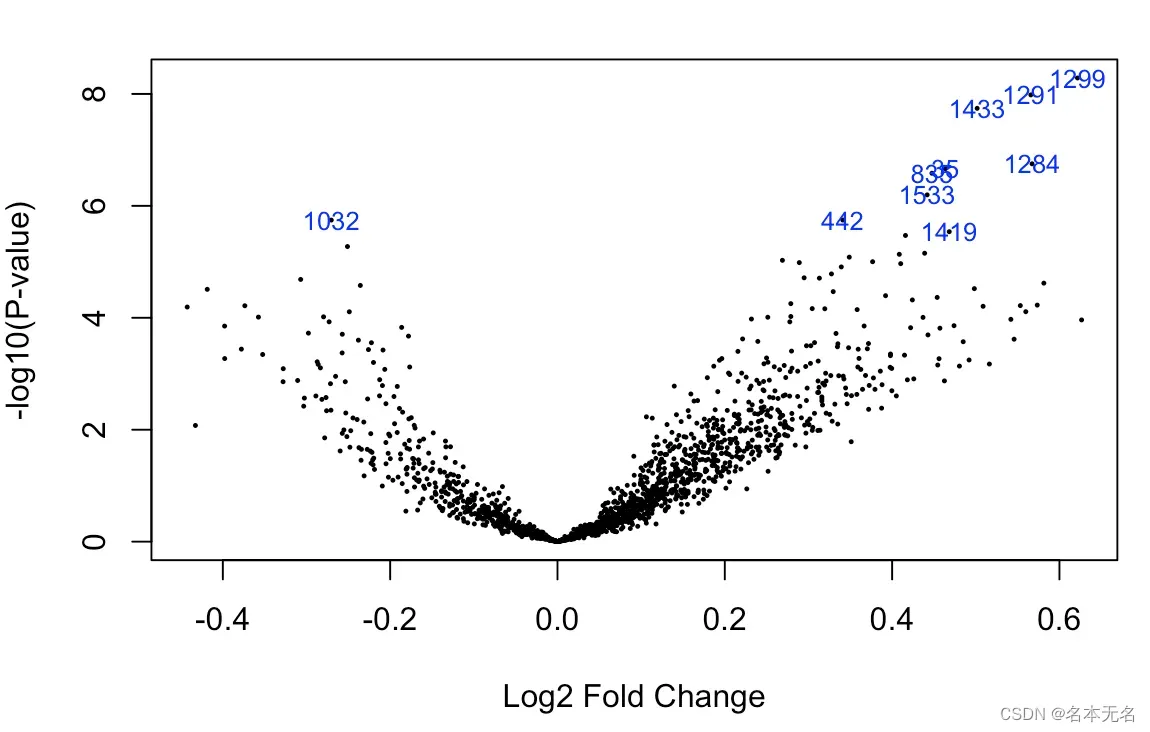
我们可以一起看下基因的火山图
dea.gene <- tcgaanalyze_dea(
mat1 = luad.exp,
mat2 = lusc.exp,
metadata = false,
pipeline = "limma",
cond1type = "luad",
cond2type = "lusc",
fdr.cut = 0.01,
logfc.cut = 1
)
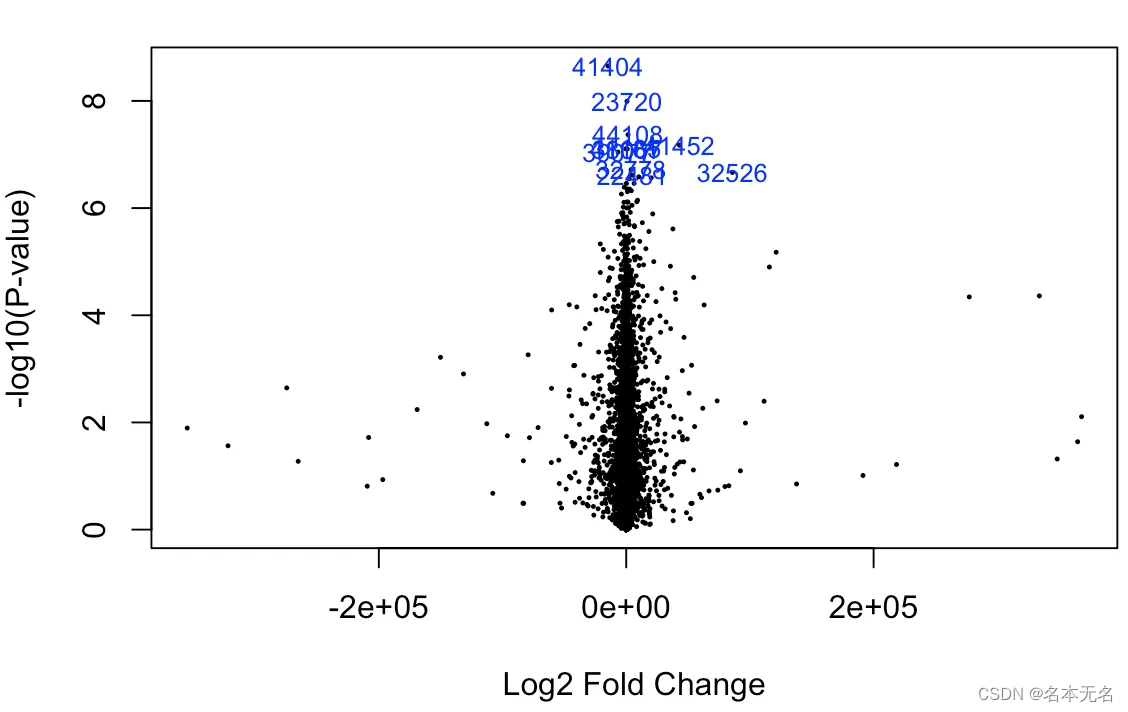
总共识别出 3157 个差异表达基因
ssgsea
single sample gene set enrichment analysis (ssgsea) 是针对单个样本进行 gsea 分析,其基因列表的排序方式和 es 的计算方式都是依赖于样本中基因的表达值,而不再是依赖基因与表型的相关度
使用方式也很简单,只要在 gsva 函数中指定 method = "ssgsea",例如
res.ssgsea <- gsva(as.matrix(gene.counts), gene.set, method = "ssgsea", kcdf = "poisson", min.sz = 10)
也可以进行差异分析
dea.ssgsea <- tcgaanalyze_dea(
mat1 = res.ssgsea[, colnames(luad.exp)],
mat2 = res.ssgsea[, colnames(lusc.exp)],
metadata = false,
pipeline = "limma",
cond1type = "luad",
cond2type = "lusc",
fdr.cut = 0.05,
logfc.cut = 0.1,
)
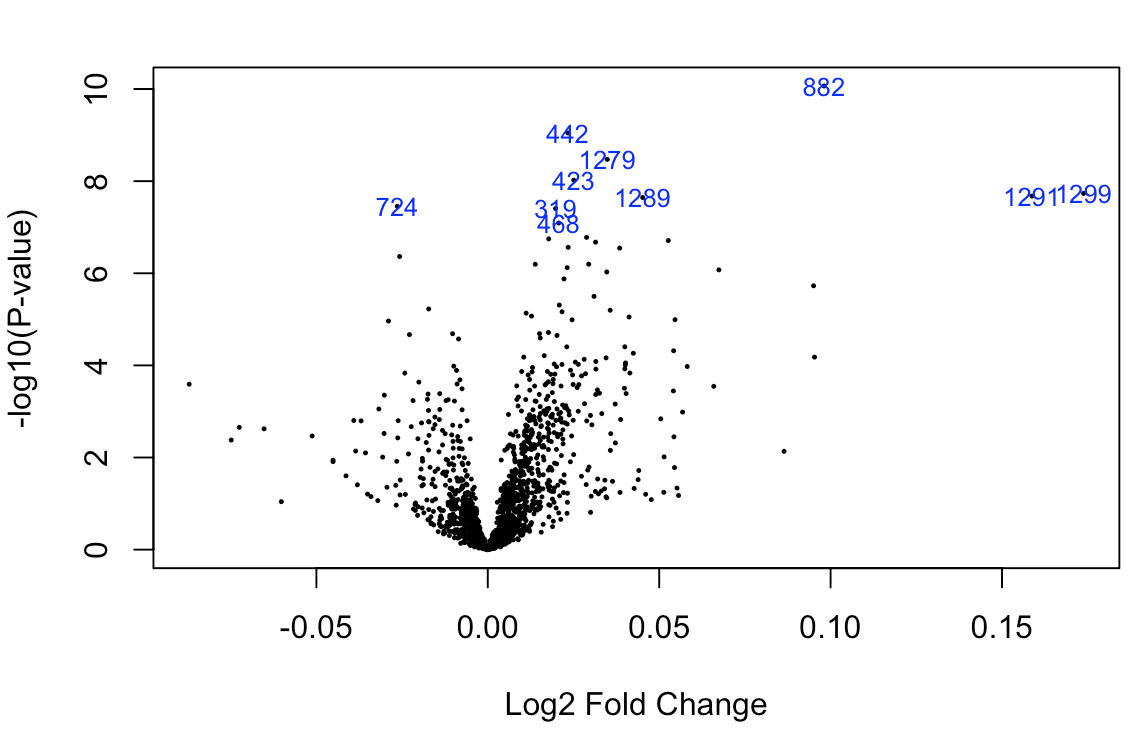
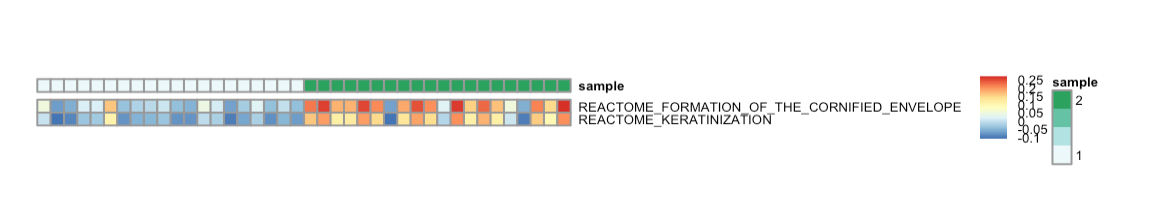
或者绘制热图
annotation_col <- data.frame(sample = rep(1:2, each = 20))
rownames(annotation_col) <- c(colnames(luad.exp), colnames(lusc.exp))
pheatmap::pheatmap(
res.ssgsea[rownames(dea.ssgsea), rownames(annotation_col)],
show_colnames = f,
# 不展示行名
cluster_rows = f,
# 不对行聚类
cluster_cols = f,
# 不对列聚类
annotation_col = annotation_col,
# 加注释
cellwidth = 5,
cellheight = 5,
# 设置单元格的宽度和高度
fontsize = 5
)
单基因富集分析
单基因富集分析并不是说拿单个基因来进行富集分析,单个基因怎么能进行富集分析呢?一个基因根本没法进行统计检验。
其实,这里说的单基因并不是拿单个基因来富集,而是基于单个基因来进行富集分析,这个“基于”,就是以单个基因为基础,向外扩展,抓取与其相关的基因,然后用这些相关的基因来进行功能富集
所以,要理解这个单基因富集分析的意思,这样一说就已经很明了了。针对单个基因我们可以做什么?
主要有两种做法:
-
定性分组:我们可以根据给定基因的表达值对样本进行分组,然后识别在两组样本之间差异表达的基因,最后用这些差异表达基因来进行功能富集
-
定量相关:通过计算其他基因与目标基因表达之间的相关性,将具有显著相关的基因作为一个集合,也可以进行富集分析
定性分组
我们以 ccdc134 基因为例,以该基因表达值的中位值来对样本进行分组
gene <- "ccdc134"
gene.exp <- gene.counts[gene,] %>%
t() %>% as.data.frame() %>%
mutate(label = ifelse(!!sym(gene) < median(!!sym(gene)), 0, 1))
group.low <- gene.counts[,gene.exp$label == 0]
group.high <- gene.counts[,gene.exp$label == 1]
识别两组样本之间的差异表达基因
degs <- tcgaanalyze_dea(
mat1 = group.low,
mat2 = group.high,
metadata = false,
pipeline = "limma",
cond1type = "ccdc134_low",
cond2type = "ccdc134_high",
fdr.cut = 0.01,
logfc.cut = 1,
)
共识别出 710 个差异表达基因
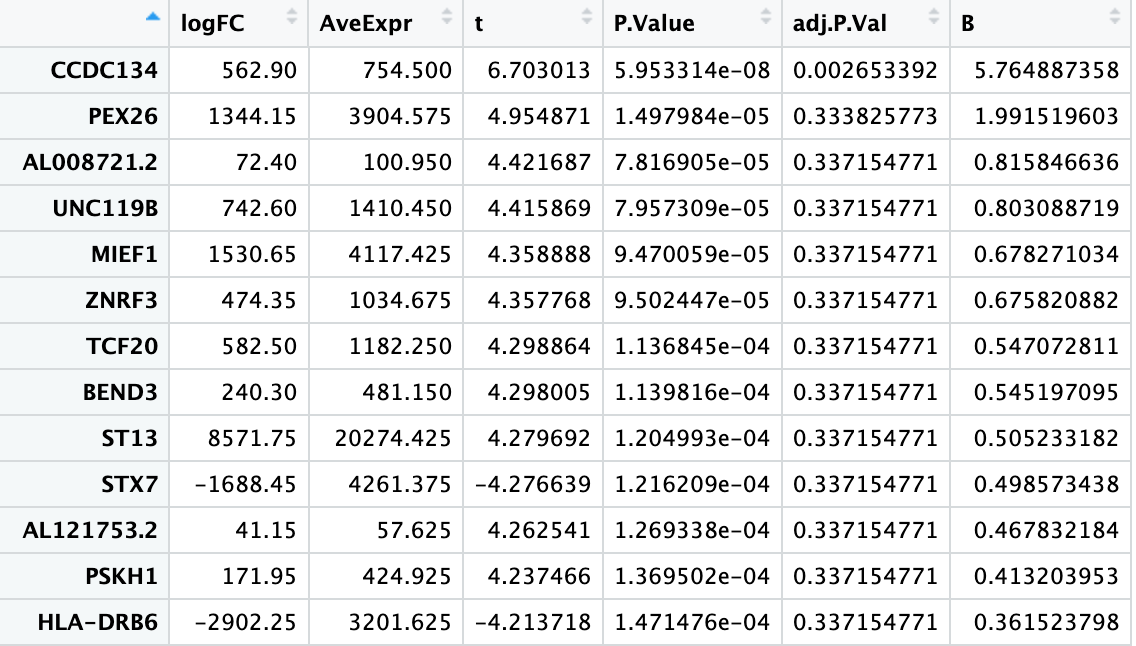
富集分析
对差异基因进行富集分析
library(clusterprofiler)
library(org.hs.eg.db)
library(enrichplot)
gene.id <- bitr(
rownames(degs), fromtype = "symbol",
totype = "entrezid",
orgdb = org.hs.eg.db
)
go <- enrichgo(
gene = gene.id$entrezid,
orgdb = org.hs.eg.db,
ont = "all",
padjustmethod = "bh",
qvaluecutoff = 0.05,
readable = t
)
dotplot(go)
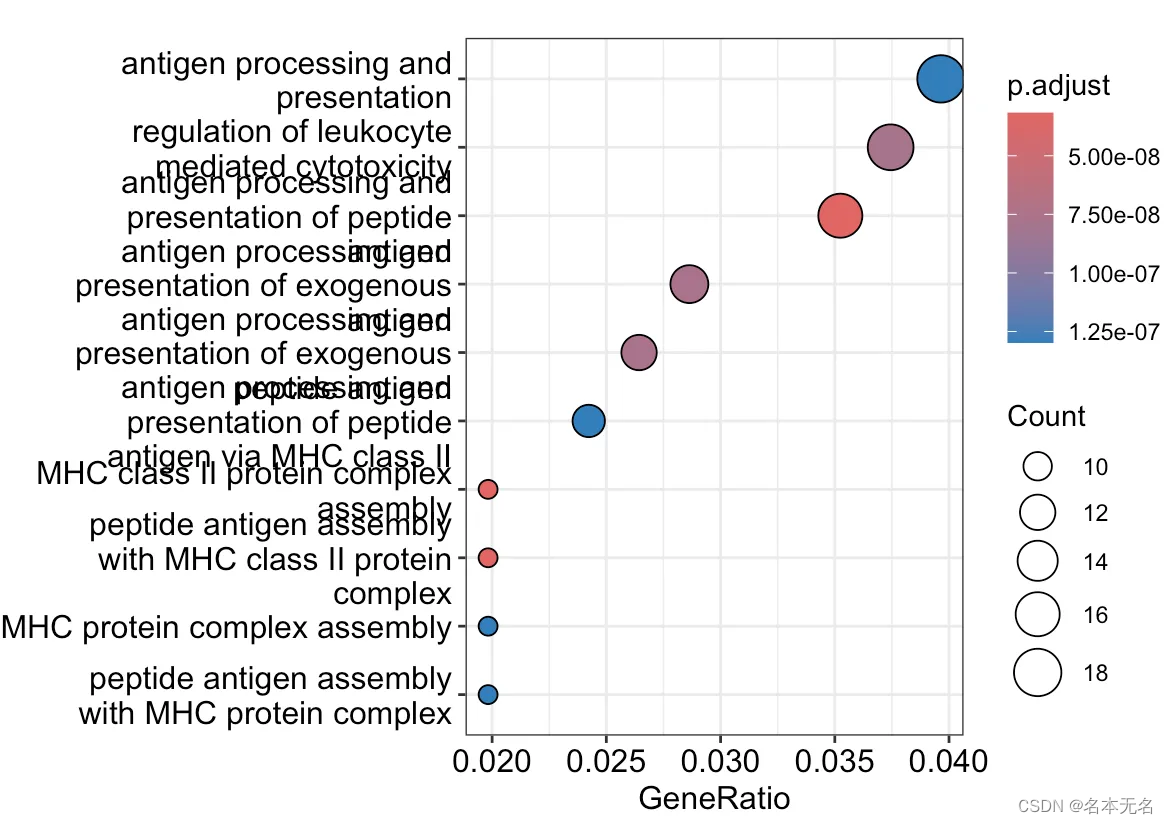
gsea 富集
gene_info <- degs %>%
rownames_to_column(var = "symbol") %>%
inner_join(., gene.id[,1:2], by = "symbol") %>%
# 必须降序
arrange(desc(logfc))
# 构造输入数据格式
genelist <- gene_info$logfc
names(genelist) <- as.character(gene_info$entrezid)
go2 <- gsego(
genelist = genelist,
orgdb = org.hs.eg.db,
ont = "all",
mingssize = 10,
maxgssize = 500,
pvaluecutoff = 0.1,
verbose = false
)
dotplot(go2)
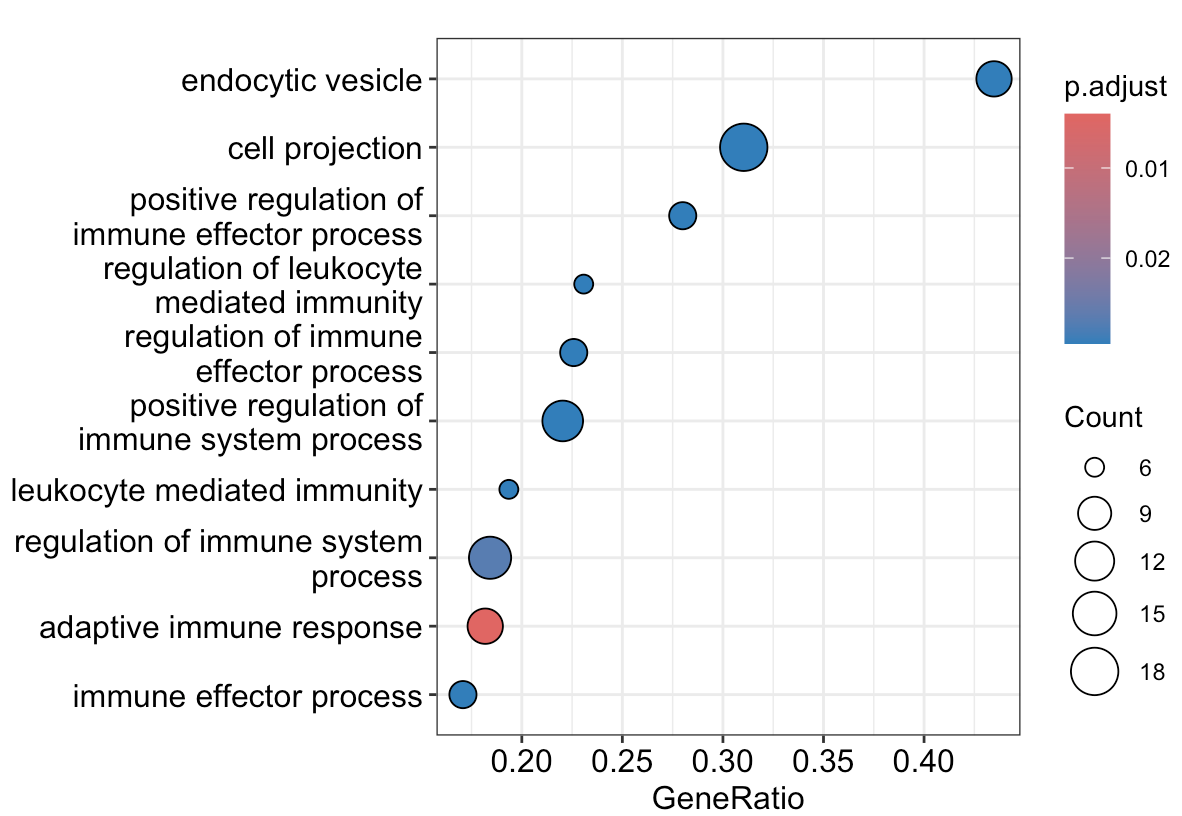
定量相关
我们计算 ccdc134 基因与其他基因的相关性,筛选出相关系数的绝对值大于 0.6 的基因,共筛选出 37 个基因。
cor.genes <- cor(t(gene.counts), as.numeric(gene.exp$ccdc134)) %>%
as.data.frame() %>%
tibble::rownames_to_column("gene") %>%
rename_with(.cols = 2, ~"corr") %>%
arrange(-corr) %>%
dplyr::filter(gene != "ccdc134")
high.cor <- cor.genes %>%
dplyr::filter(abs(corr) > 0.6)
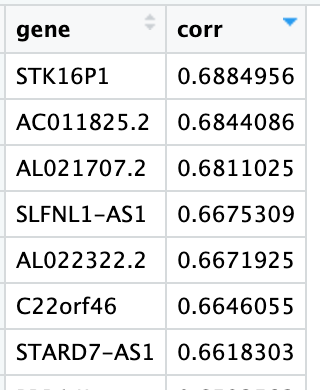
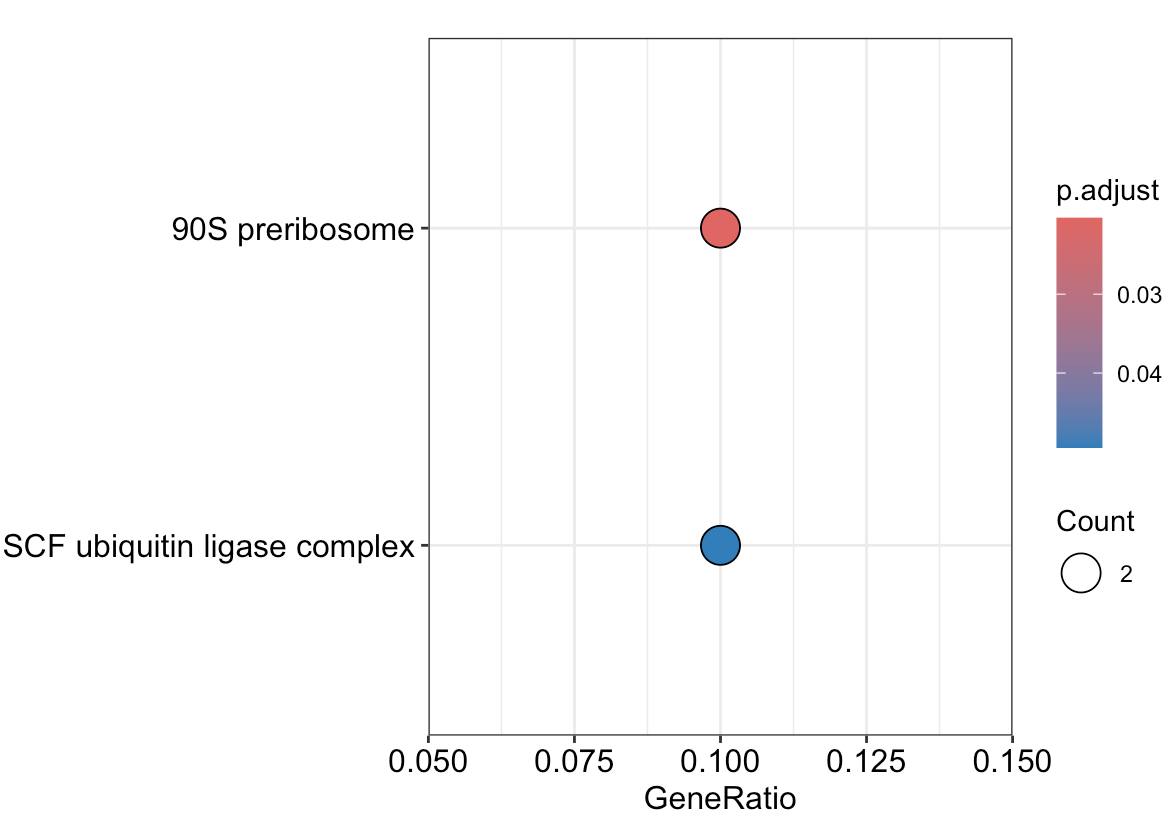
富集分析
使用高度相关的基因进行富集分析
go <- enrichgo(
gene = high.cor$gene,
orgdb = org.hs.eg.db,
keytype = "symbol",
ont = "all",
padjustmethod = "bh",
qvaluecutoff = 0.05,
readable = t
)
dotplot(go)
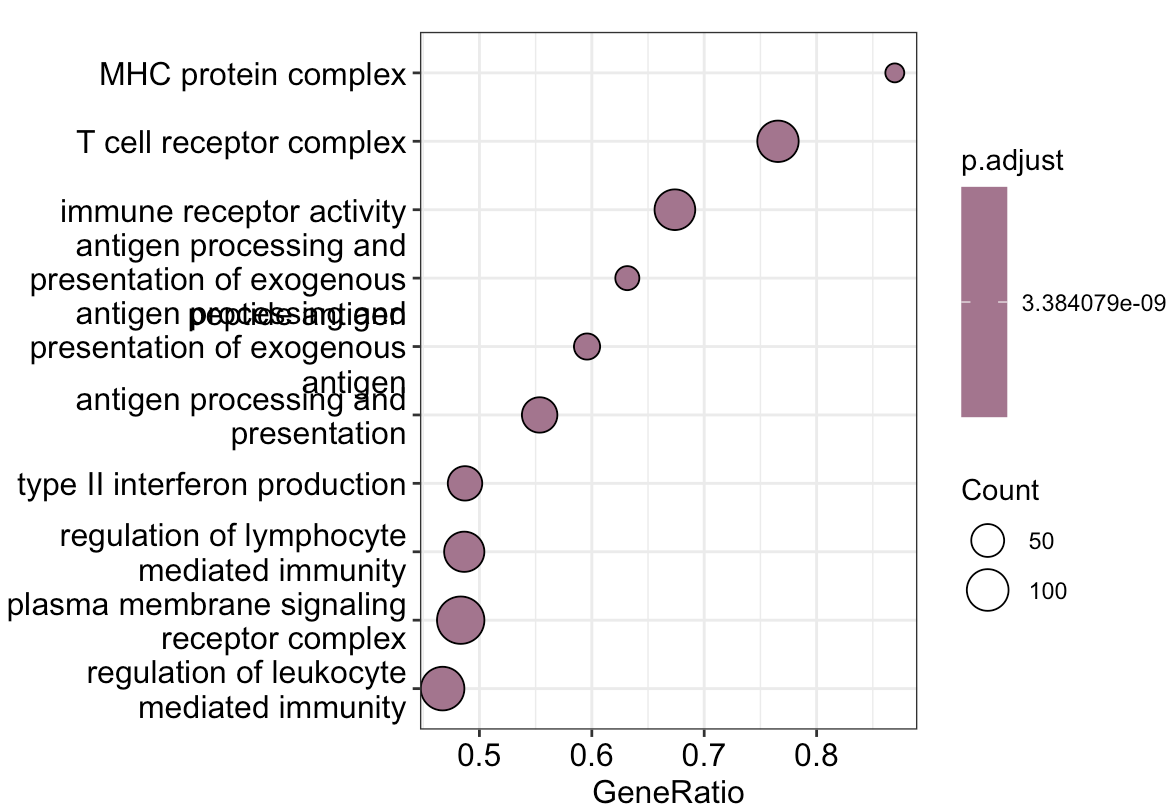
根据相关性的大小排序,然后进行 gsea 分析
# 构造输入数据格式
genelist <- cor.genes$corr
names(genelist) <- as.character(cor.genes$gene)
go2 <- gsego(
genelist = genelist,
orgdb = org.hs.eg.db,
keytype = "symbol",
ont = "all",
mingssize = 10,
maxgssize = 500,
pvaluecutoff = 0.1,
verbose = false
)
dotplot(go2)




发表评论