零基础入门转录组数据分析——数据处理(geo数据库——高通量测序数据)
目录
您首先需要了解本贴是完全免费按实际案例分享基础知识和全部代码,希望能帮助到初学的各位更快入门,但是 尊重创作和知识才会有不断高质量的内容输出 ,如果阅读到最后觉得本贴确实对自己有帮助,希望广大学习者能够花点自己的小钱支持一下作者创作(条件允许的话一杯奶茶钱即可),感谢大家的支持~~~~~~ ^_^ !!!
注:当然这个并不是强制的哦,大家也可以白嫖~~,只是一点点小的期盼!!!
祝大家能够开心学习,轻松学习,在学习的路上少一些坎坷~~~
geo数据库全称gene expression omnibus,是由美国国立生物技术信息中心ncbi创建并维护的基因表达数据库。它创建于2000年,收录了世界各国研究机构提交的高通量基因表达数据,也就是说只要是目前已经发表的论文,论文中涉及到的基因表达检测的数据都可以通过这个数据库中找到。
并且geo网站这个网站作为各种高通量实验数据的公共存储库。这些数据包括基于单通道和双通道微阵列的实验,检测mrna,基因组dna和蛋白质丰度,以及非阵列技术,如基因表达系列分析(sage),质谱蛋白质组学数据和高通量测序数据。可以按照文献数据集编号等众多形式进行检索。但是在这篇教程中仅介绍如何从geo网站上根据数据集编号下载所需要的geo数据集,并且下载后在r中对数据集进一步处理成后续分析所要的形式。
本项目以妊娠期糖尿病gse154414数据集(高通量测序数据)作为展示
选用的数据库是geo。
实验分组:疾病组,对照组。
我这里使用的r版本是4.2.2
要用到的r包:tidyverse,geoquery
1. 数据集获取
首先进入geo网站官网(如下图所示),在检索位置输入数据集编号,点击箭头指向的位置进一步运行搜索。
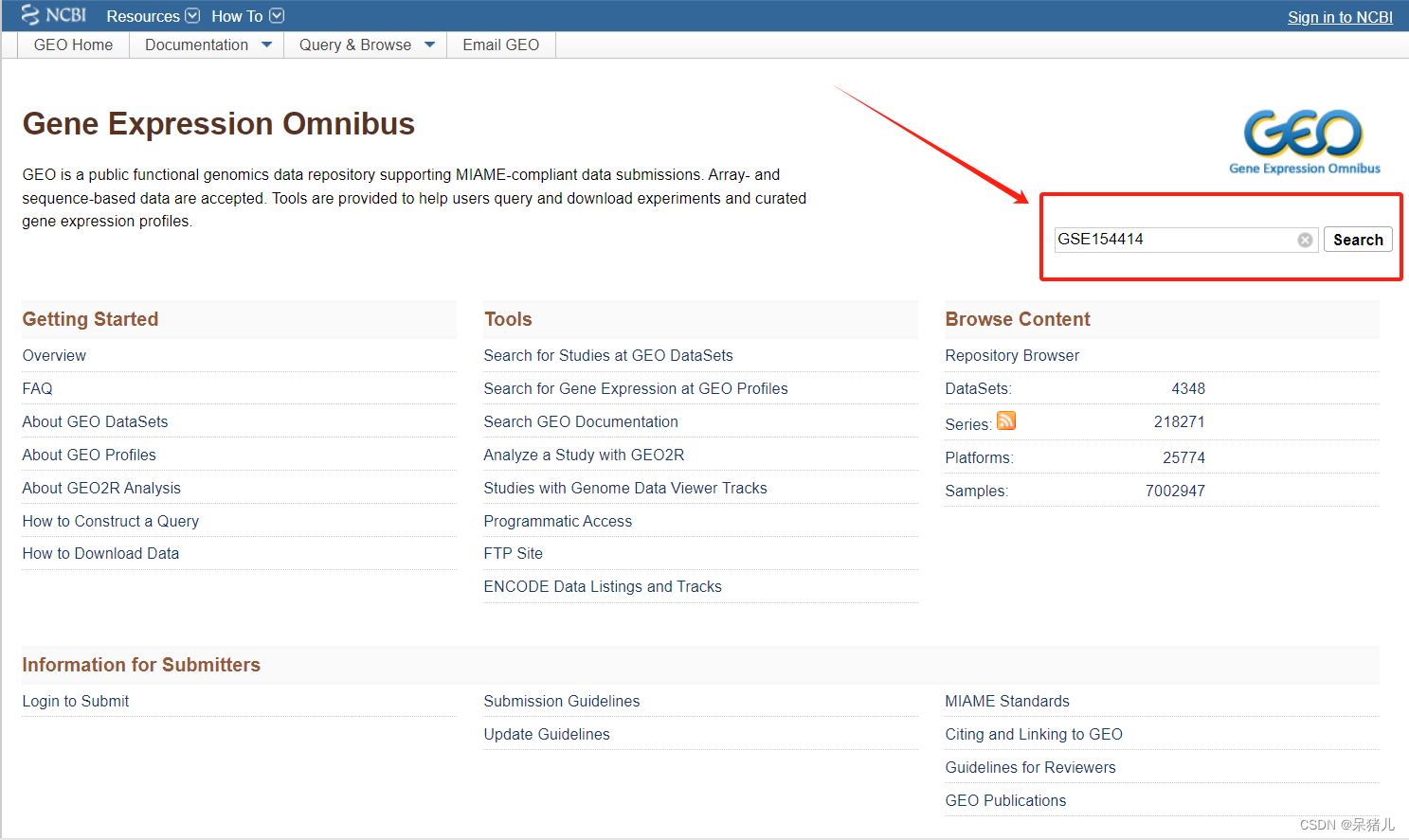
搜索之后会弹出如下界面:首先需要检查物种类型(homo sapiens),之后查看数据集的类型是否是高通量测序/芯片数据,我这里是高通量测序数据(expression profiling by high throughput sequencing),页面往下拉。
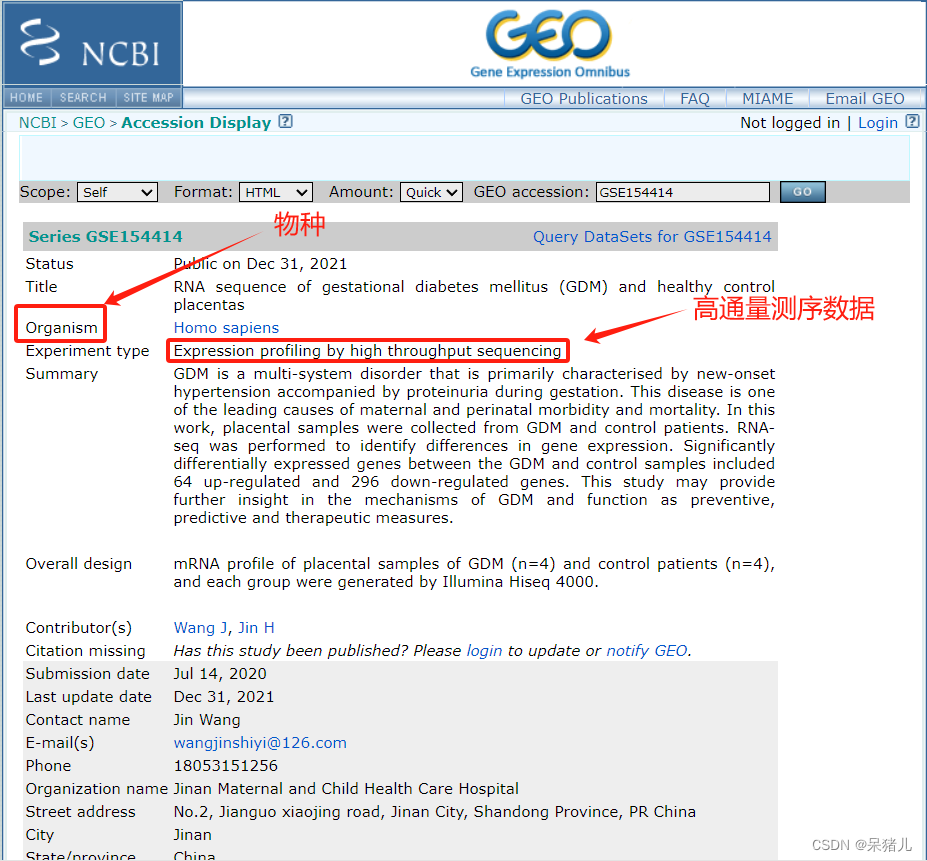
如下图所示:包含了该数据集对应的注释文件gpl20301,并且还列出来了数据集中包含的样本。
注:但是注释信息可以不用过多的关注,因为后续分析用不到,样本数量可以大致瞅一眼

到此对于该数据集已经有了初步了解(实际上就是看是不是高通量测序数据),如果是高通量测序数据就按照下面的操作进行,如果是芯片数据,可以参考之前的教程零基础入门转录组分析——数据处理(geo数据库——芯片数据)
2. 数据处理(rstudio)
rm(list = ls()) # 删除工作空间中所有的对象
setwd('/xx/xx/xx') # 设置工作路径
if(!dir.exists('./00_rawdata')){
dir.create('./00_rawdata')
} # 判断该工作路径下是否存在名为00_rawdata的文件夹,如果不存在则创建,如果存在则pass
setwd('./00_rawdata/') # 设置路径到刚才新建的00_rawdata下
加载包:
library(geoquery)
library(tidyverse)
标注一下需要下载的数据集编号,并且在当前00_rawdata文件夹下创建一个名为gse154414的文件夹(这是为了方便管理,如果不单独创建文件夹,数据集很多的话,就会显得很乱)
geo_data <- 'gse154414'
if(!dir.exists(paste0(geo_data))){
dir.create(paste0(geo_data))
}
setwd(paste0(geo_data))
获取数据集:
- 第一行指令是ncbi存放数据的地址
- 第二行是拼接网址为gse154414原始count存放地址(细心的小伙伴或许已经观察到里面acc=xxx,file=xxx这里就是我们的gse154414,这里其实用到了一点爬虫的思路!)
- 第三行指令是获取gse154414的原始count数据,等待下载完成
# load raw count of data_set
urld <- "https://www.ncbi.nlm.nih.gov/geo/download/?format=file&type=rnaseq_counts"
path <- paste(urld, "acc=gse154414", "file=gse154414_raw_counts_grch38.p13_ncbi.tsv.gz", sep="&");
tbl <- as.matrix(data.table::fread(path, header=t, colclasses="integer"), rownames="geneid")

下载完成后,优先转换成数据框格式并保存(这是因为有的数据集非常大,下载要好久,有时候碰到网不好的时候还容易下载报错,各种失败,所以要及时保存)
expr <- as.data.frame(tbl)
write.csv(expr, paste0(geo_data, '_raw_count.csv'))
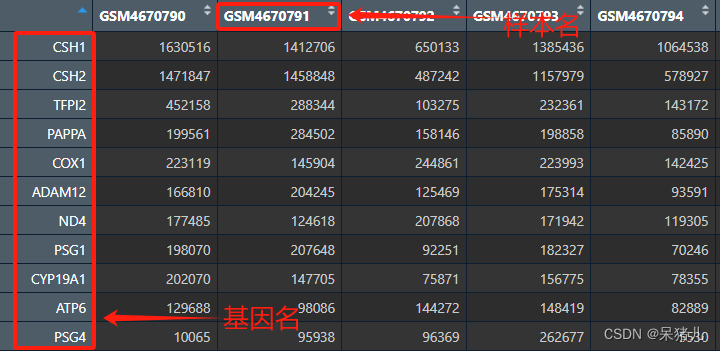
可以先看一下下载好的这个名为expr 的数据长啥样,如下图所示,行为基因的id,列为样本名,表中的那些整数就是raw_count。
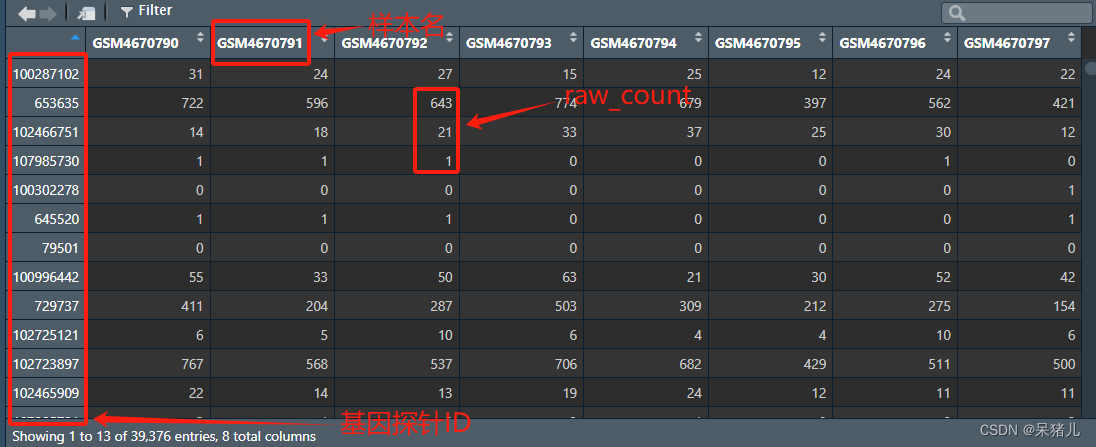
接下来就是将这个表达矩阵的行名转换成我们平常看到的基因symbol,要不然谁知道这些是个什么东西(>_>)
下载基因注释文件(这一步是用官方的注释文件)同样也是用的上述相似的代码获取,如下所示:
# load gene annotations
apath <- paste(urld, "type=rnaseq_counts", "file=human.grch38.p13.annot.tsv.gz", sep="&")
annot <- data.table::fread(apath, header=t, quote="", stringsasfactors=f, data.table=f)
rownames(annot) <- annot$geneid
write.csv(annot, 'gene_annoation.csv')
同样打开下载好的这个名为annot的基因注释文件,看看都是些什么,如下图所示:
- 第一列就是与表达矩阵行名一样的基因id
- 第二列就是基因的symbol(也就是咱们平常见的基因名)
- 第三列就是关于基因的简要描述
- 第五列是基因类型(有的基因是编码基因,有的是非编码基因)
- 后面还有基因对应的ensemblgeneid,基因长度,位于染色体的位置,以及是否是正链/负链等信息(可以按需从这个注释表中获取基因的相应信息)
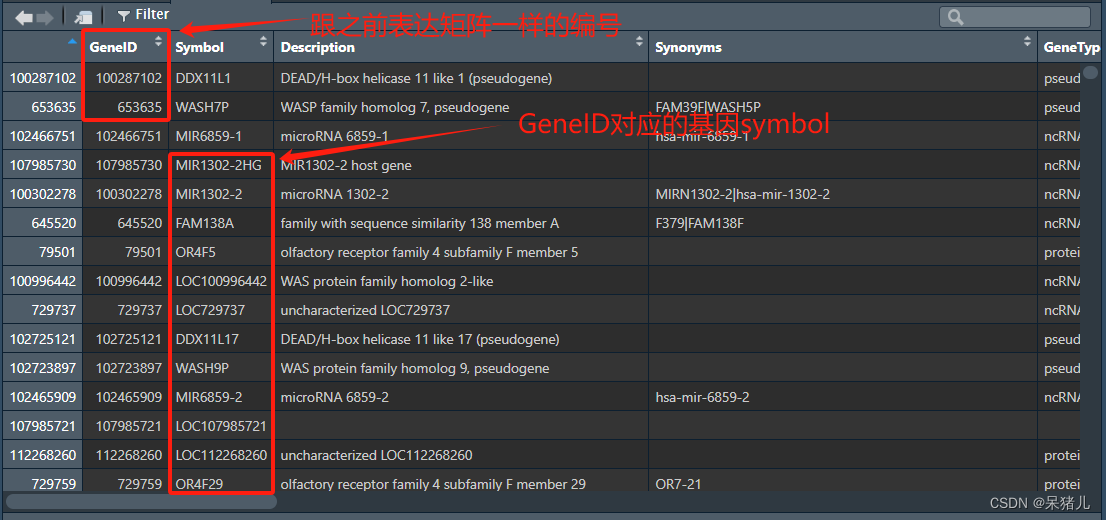
注意:这个注释文件适用于基本所有geo高通量数据(因此只需下载一次即可,做好保存,遇到其他geo高通量数据可以直接调用,不用重新下载)
接下来将这个基因注释表调整成后续需要用的格式(我这里第一步筛选了一下编码基因,如果不需要筛选的小伙伴可以把这一步注释掉)
table(annot$genetype) # 查看genetype都有哪些类型
annot <- annot[annot$genetype == 'protein-coding', ] # 选择编码基因
colnames(annot) # 查看列名
probe2symbol <- dplyr::select(annot, 'geneid', 'symbol') # 选择基因id和基因symbol这两列
probe2symbol <- filter(probe2symbol, `symbol` != '') # 去掉symbol中为空的行
names(probe2symbol) <- c('id', 'symbol') # 给geneid和symbol两列名重命名为id和symbol
probe2symbol <- na.omit(probe2symbol) # 去除为na的行
最终处理好的probe2symbol 如下图所示,第一列是基因id,第二列为基因symbol
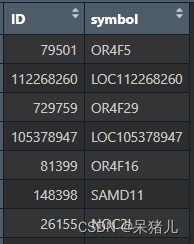
接下来要将前面的expr表达矩阵和处理好的probe2symbol 联系到一起(说白了就是给expr表达矩阵的行名注释成我们平常看到的基因名,并且加个去重处理)
expr$id <- rownames(expr) # 给expr定义一个名为id的新列,每一行的值为expr的行名
dat <- expr # 将expr赋值给dat
dat$id <- as.character(dat$id) # 将dat的id列转成字符串型
probe2symbol$id <- as.character(probe2symbol$id)
dat <- dat %>%
merge(probe2symbol, by='id')%>%
dplyr::select(-id)%>% ## 去除多余信息
dplyr::select(symbol, everything())%>% ## 重新排列
mutate(rowmean = rowmeans(.[grep('gsm', names(.))]))%>% ## 求出平均数
arrange(desc(rowmean))%>% ## 把表达量的平均值从大到小排序
distinct(symbol, .keep_all = t)%>% ## symbol留下第一个
dplyr::select(-rowmean)%>% ## 反向选择去除rowmean这一列
tibble::column_to_rownames(colnames(.)[1]) ## 把第一列变成行名并删除
最终处理好的dat 如下图所示,列名为样本名,行名为平常常见的基因名(可以看出来这个和没处理前expr的最大区别就是:基因id转成了熟悉的基因名)。
接下来获取样本的临床信息表,其中getgeo函数是从geo数据库中获取数据集的相关信息(包括样本的临床信息和表达矩阵等,但是由于这个数据集是高通量测序数据,因此表达矩阵不在这里)。
gset <- getgeo(geo_data ,
destdir = '.',
gsematrix = t,
getgpl = f)
如下图所示gset是一个列表,phenodata里面就含有样本的临床信息。

通过pdata函数获取样本临床信息
a <- gset[[1]]
pd <- pdata(a)
pd文件如下图所示每一行是一个样本,每一列为样本的不同临床信息,包括但不局限于:样本名,肿瘤分期,肿瘤分级,生存信息,样本类型等。
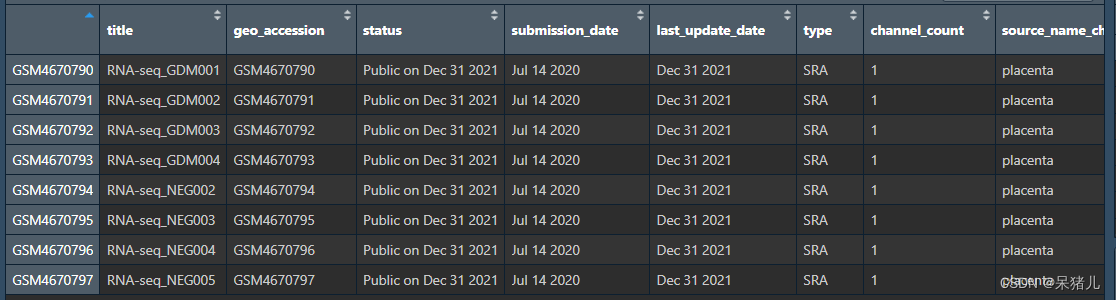
注:这个样本信息表中每一列命名不是规范的,如果想要获取自己需要的哪列临床信息(例如:样本类型——是否是癌,生存信息等),需要一一去查看对应的列中信息都是什么
在这里我们需要的是样本类型信息(用于做分组),这个信息存储在characteristics_ch1.2这列中
table(pd$characteristics_ch1.2)
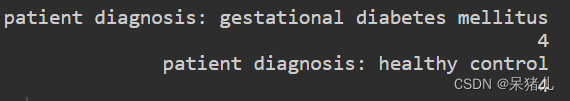
table之后可以看到这列中包含两种类型,每种类型有4个样本:
- patient diagnosis: gestational diabetes mellitus(患者诊断:妊娠期糖尿病)
- patient diagnosis: healthy control(患者诊断:健康对照)
接下来就可以根据这列信息做成我们自己想要的分组形式(即:一列是样本id,另一列是分组情况),比如:将patient diagnosis: gestational diabetes mellitus作为疾病组,patient diagnosis: healthy control作为对照组
group <- data.frame(sample = pd$geo_accession, group = pd$characteristics_ch1.2) # 创建一个数据框,包含两列:sample和group
table(group$group)
group$group <- ifelse(group$group == 'patient diagnosis: healthy control', 'control', 'disease') # 判断,如果group列中值为patient diagnosis: healthy control,则重命名为control,否则为disease
table(group$group)
group <- group[order(group$group), ]
group文件如下图所示:第一列是样本id,第二列是分组类型。

到这一步基本表达矩阵和样本分组信息表已经处理好了,通过 xx %in% xx筛选样本(这一步是因为有的数据集中不仅有癌与癌旁,还有可能会有别的类型的样本,在前面做完筛选后和处理好的表达矩阵样本数量就不一样了,因此要保持一致)后就可以保存了。
colnames(dat)
group$sample
dat <- dat[, colnames(dat) %in% group$sample] # 筛选group中存在的样本
dat <- na.omit(dat)
group <- group[group$sample %in% colnames(dat), ] # 筛选dat中存在的样本
write.csv(dat, file = paste0('dat.', geo_data, '_count.csv'))
write.csv(group, file = paste0('group.', geo_data, '.csv'), row.names = f)
到这一步数据集原始表达矩阵(raw_count)就已经处理完了,这个表达矩阵可以接下来通过deseq2进行差异分析。
注:raw_count仅适用于做差异分析,如果是做其他分析,则需要对raw_count进行标准化处理,转成fpkm后才能用于其他类型的分析(例如:wgcna,生存分析等)
接下来就是对raw_count做标准化处理,将其转换成fpkm形式。
3. 数据标准化(rstudio)
关于fpkm的解释,可以参考rpkm、fpkm 和 tpm,解释清楚这篇文章(简单说就是由于存在测序深度不同/基因长度不同的情况,所以原始count并不能真实反映基因表达量,有的基因长度较长,匹配到的reads会非常多,因此会根据基因长度做个校正)
首先要从前面annot文件中获取基因的长度
colnames(annot)
gene_length <- dplyr::select(annot, symbol, length)
gene_length <- gene_length[gene_length$symbol %in% rownames(dat), ]
定义一个名为counttofpkm 的计算函数:
counttofpkm <- function(counts, efflen){
n <- sum(counts)
exp( log(counts) + log(1e9) - log(efflen) - log(n) )
}
通过apply函数和之前自定义的counttofpkm 函数对表达矩阵进行fpkm转换
dat <- dat[gene_length$symbol, ]
expr_fpkm <- apply(dat, 2, counttofpkm, efflen = gene_length$length)
expr_fpkm就是经过fpkm转换的数据集(但是目前还不能直接用,是因为除了差异分析之外,基本所有的分析点用到的数据集都是log2()处理的,这是因为纯fpkm值有的会很大,不同分析点在计算的时候可能会由于个别值特别大会导致较大的误差,因此要通过log2处理。)
接下来通过geo官方的判定公式,如果logc的值为true则对expr_fpkm数据集进行log2处理,如果为false则不处理
qx <- as.numeric(quantile(expr_fpkm, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=t))
logc <- (qx[5] > 100) ||
(qx[6]-qx[1] > 50 && qx[2] > 0) ||
(qx[2] > 0 && qx[2] < 1 && qx[4] > 1 && qx[4] < 2)
if (logc) {
expr_fpkm[which(expr_fpkm <= 0)] <- nan
expr_fpkm <- log2(expr_fpkm + 1)
print("log2 transform finished")
}else{
print("log2 transform not needed")
}
注:这里需要特别注意一点的是这个判定函数并不是万能的,有些数据集在这一步判定的时候为false,但是里面的fpkm值都很大,像这种特殊情况的就不能用判定了,可以直接对其做log2处理
最后将处理好的数据集保存即可
expr_fpkm <- data.frame(expr_fpkm)
expr_fpkm <- na.omit(expr_fpkm)
write.csv(expr_fpkm, file = paste0('dat.', geo_data, '_fpkm.csv'))
补充内容:
- 为什么要进行log2处理:一般数据集的表达值范围都是非常大的,从0到25000不等。将这些数据绘制密度分布后,一般呈现右偏,即大部分信号都是在左侧,右侧拖个长长的尾巴(偏态分布),不利于研究,而经过log2转化后,数据更加集中,更加接近正态分布,更方便套用正态分布那一套理论进行后续分析。
以上就是geo高通量测序数据下载及处理的所有过程,如果有什么需要补充或不懂的地方,大家可以私聊我或者在下方评论。
如果觉得本教程对你有所帮助,点赞关注不迷路!!!
与教程配套的原始数据+代码+处理好的数据见零基础入门转录组分析——数据处理(geo数据库——高通量测序数据)配套资源
注:配套资源只要改个路径就能运行,本人已检测过可以跑通,请放心食用,食用过程遇到问题,可先自行百度,实在解决不了可以私信
- 目录部分跳转链接:


发表评论